

Detecting cell-of-origin and cancer-specific methylation features of cell-free DNA from Nanopore sequencing
- PMID: 35841107
- PMCID: PMC9283844
- DOI: 10.1186/s13059-022-02710-1
Detecting cell-of-origin and cancer-specific methylation features of cell-free DNA from Nanopore sequencing
Abstract
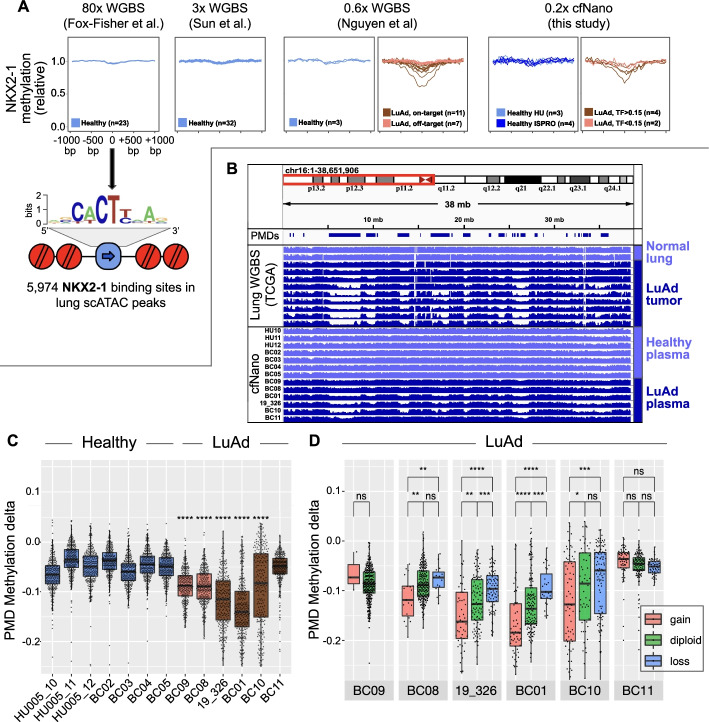
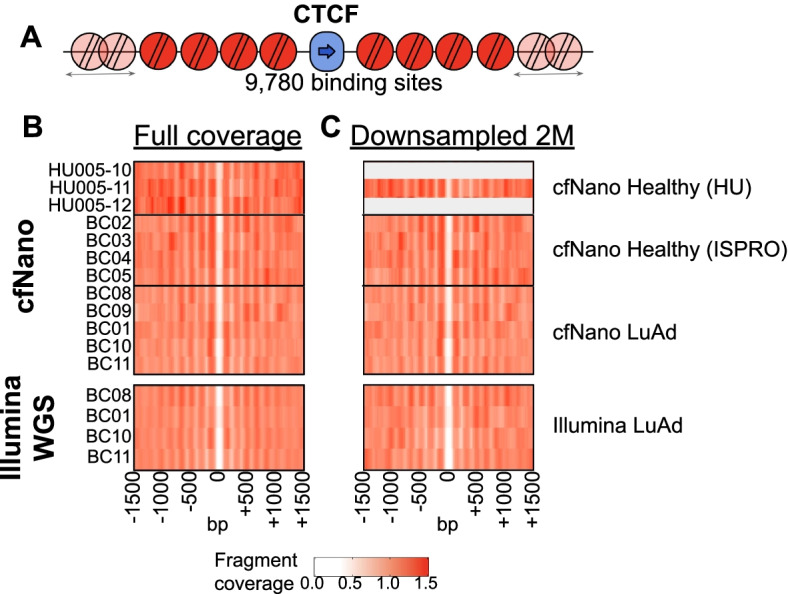
The Oxford Nanopore (ONT) platform provides portable and rapid genome sequencing, and its ability to natively profile DNA methylation without complex sample processing is attractive for point-of-care real-time sequencing. We recently demonstrated ONT shallow whole-genome sequencing to detect copy number alterations (CNAs) from the circulating tumor DNA (ctDNA) of cancer patients. Here, we show that cell type and cancer-specific methylation changes can also be detected, as well as cancer-associated fragmentation signatures. This feasibility study suggests that ONT shallow WGS could be a powerful tool for liquid biopsy.
© 2022. The Author(s).
Conflict of interest statement
BPB, EK, SO, FM, and SGC are inventors on IP filings of this work by the Yissum Research Development Company of The Hebrew University of Jerusalem Ltd. BPB and SO receive research funding from Volition Belgium Rx for an unrelated study.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
