

Necroptosis and Neuroinflammation in Retinal Degeneration
- PMID: 35844208
- PMCID: PMC9277228
- DOI: 10.3389/fnins.2022.911430
Necroptosis and Neuroinflammation in Retinal Degeneration
Abstract
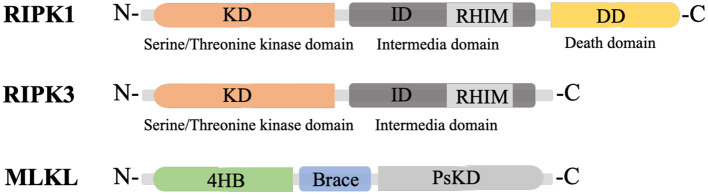
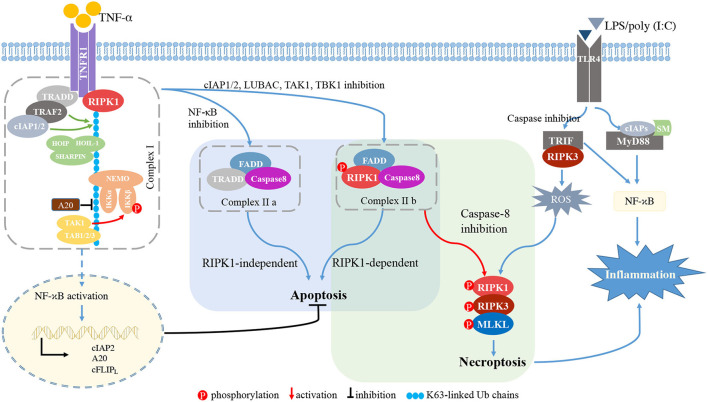
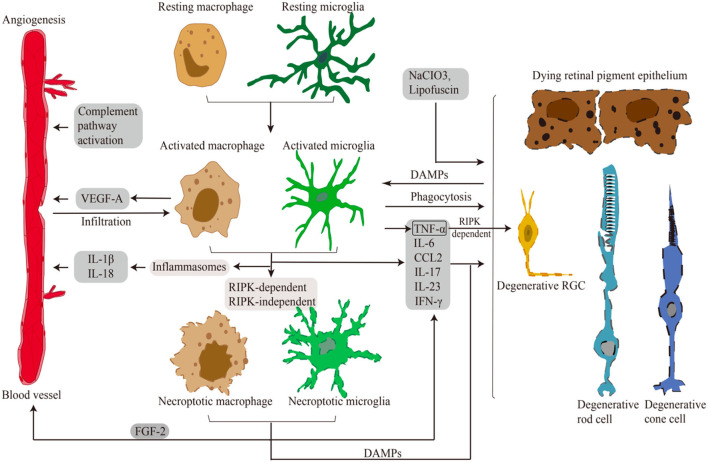
Necroptosis mediates the chronic inflammatory phenotype in neurodegeneration. Receptor-interacting protein kinase (RIPK) plays a pivotal role in the induction of necroptosis in various cell types, including microglia, and it is implicated in diverse neurodegenerative diseases in the central nervous system and the retina. Targeting RIPK has been proven beneficial for alleviating both neuroinflammation and degeneration in basic/preclinical studies. In this review, we discuss the role of necroptosis in retinal degeneration, including (1) the molecular pathways involving RIPK, (2) RIPK-dependent microglial activation and necroptosis, and (3) the interactions between necroptosis and retinal neuroinflammation/degeneration. This review will contribute to a renewed focus on neuroinflammation induced by necroptosis and to the development of anti-RIPK drugs against retinal degeneration.
Keywords: RIPK; microglia; necroptosis; neuroinflammation; retinal degeneration.
Copyright © 2022 Tao, Murakami, Vavvas and Sonoda.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Necroptosis in microglia contributes to neuroinflammation and retinal degeneration through TLR4 activation.Cell Death Differ. 2018 Jan;25(1):180-189. doi: 10.1038/cdd.2017.141. Epub 2017 Sep 8. Cell Death Differ. 2018. PMID: 28885615 Free PMC article.
-
Gallic Acid Attenuated LPS-Induced Neuroinflammation: Protein Aggregation and Necroptosis.Mol Neurobiol. 2020 Jan;57(1):96-104. doi: 10.1007/s12035-019-01759-7. Epub 2019 Dec 12. Mol Neurobiol. 2020. PMID: 31832973
-
Necroptosis: A Novel Pathway in Neuroinflammation.Front Pharmacol. 2021 Jul 12;12:701564. doi: 10.3389/fphar.2021.701564. eCollection 2021. Front Pharmacol. 2021. PMID: 34322024 Free PMC article. Review.
-
Loss of Microglial Parkin Inhibits Necroptosis and Contributes to Neuroinflammation.Mol Neurobiol. 2019 Apr;56(4):2990-3004. doi: 10.1007/s12035-018-1264-9. Epub 2018 Aug 3. Mol Neurobiol. 2019. PMID: 30074231
-
Microglia: Key Players in Retinal Ageing and Neurodegeneration.Front Cell Neurosci. 2022 Mar 17;16:804782. doi: 10.3389/fncel.2022.804782. eCollection 2022. Front Cell Neurosci. 2022. PMID: 35370560 Free PMC article. Review.
Cited by
-
Artificial intelligence-enabled discovery of a RIPK3 inhibitor with neuroprotective effects in an acute glaucoma mouse model.Chin Med J (Engl). 2025 Jan 20;138(2):172-184. doi: 10.1097/CM9.0000000000003387. Epub 2024 Dec 23. Chin Med J (Engl). 2025. PMID: 39719694 Free PMC article.
-
Quantification of Photoreceptors' Changes in a Diabetic Retinopathy Model with Two-Photon Imaging Microscopy.Int J Mol Sci. 2024 Aug 11;25(16):8756. doi: 10.3390/ijms25168756. Int J Mol Sci. 2024. PMID: 39201444 Free PMC article.
-
Modeling inducible neuropathologies of the retina with differential phenotypes in organoids.Front Cell Neurosci. 2023 May 5;17:1106287. doi: 10.3389/fncel.2023.1106287. eCollection 2023. Front Cell Neurosci. 2023. PMID: 37213216 Free PMC article.
-
Semaglutide alleviates early brain injury following subarachnoid hemorrhage by suppressing ferroptosis and neuroinflammation via SIRT1 pathway.Am J Transl Res. 2024 Apr 15;16(4):1102-1117. doi: 10.62347/IZGJ1332. eCollection 2024. Am J Transl Res. 2024. PMID: 38715815 Free PMC article.
-
Combination of machine learning-based bulk and single-cell genomics reveals necroptosis-related molecular subtypes and immunological features in autism spectrum disorder.Front Immunol. 2023 Apr 24;14:1139420. doi: 10.3389/fimmu.2023.1139420. eCollection 2023. Front Immunol. 2023. PMID: 37168851 Free PMC article.
References
Publication types
LinkOut - more resources
Full Text Sources