

RNA-SSNV: A Reliable Somatic Single Nucleotide Variant Identification Framework for Bulk RNA-Seq Data
- PMID: 35846154
- PMCID: PMC9279659
- DOI: 10.3389/fgene.2022.865313
RNA-SSNV: A Reliable Somatic Single Nucleotide Variant Identification Framework for Bulk RNA-Seq Data
Abstract
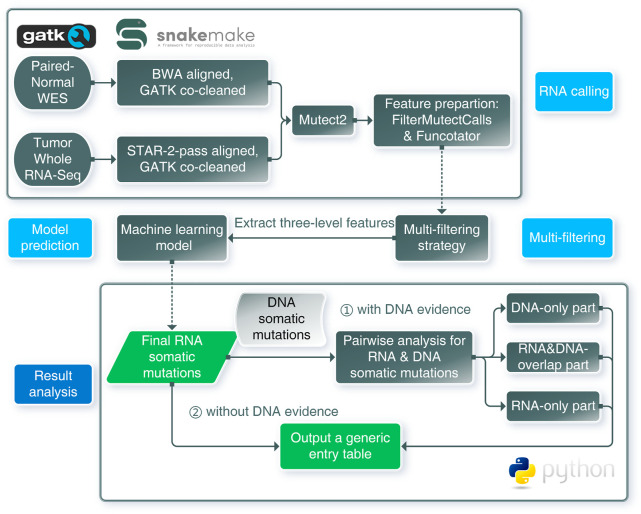
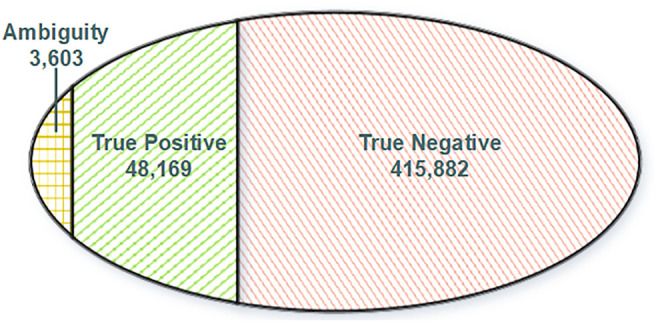
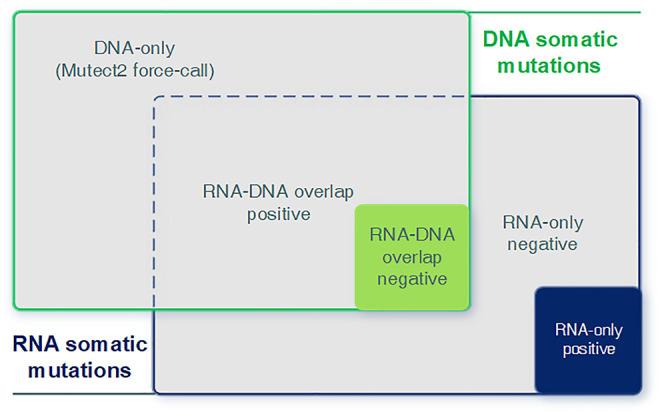
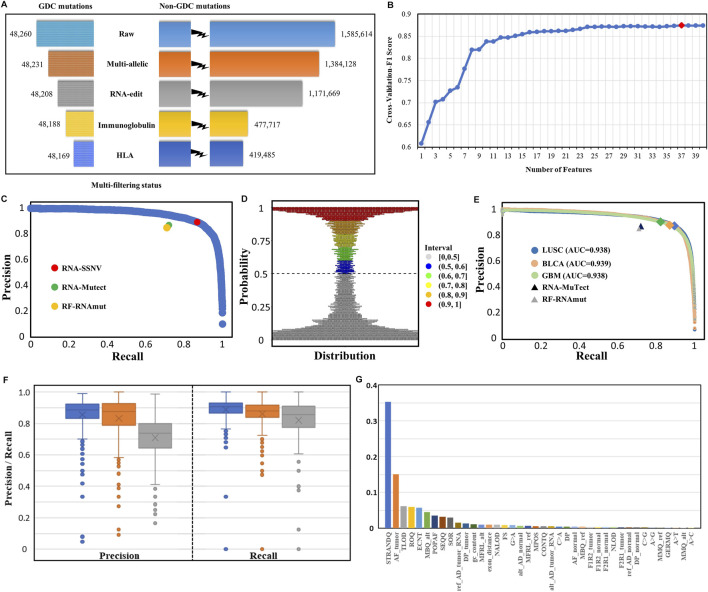
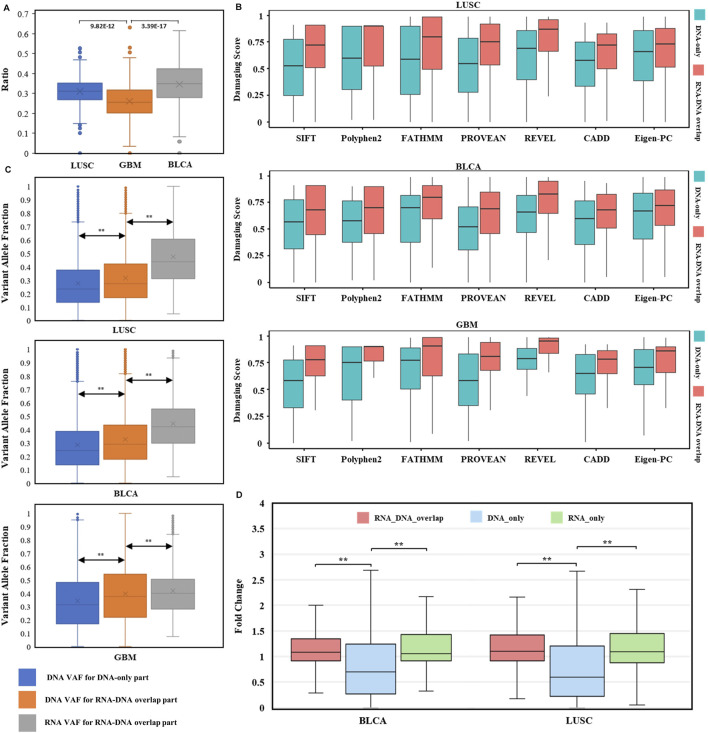
The usage of expressed somatic mutations may have a unique advantage in identifying active cancer driver mutations. However, accurately calling mutations from RNA-seq data is difficult due to confounding factors such as RNA-editing, reverse transcription, and gap alignment. In the present study, we proposed a framework (named RNA-SSNV, https://github.com/pmglab/RNA-SSNV) to call somatic single nucleotide variants (SSNV) from tumor bulk RNA-seq data. Based on a comprehensive multi-filtering strategy and a machine-learning classification model trained with comprehensively curated features, RNA-SSNV achieved the best precision-recall rate (0.880-0.884) in a testing dataset and robustly retained 0.94 AUC for the precision-recall curve in three validation adult-based TCGA (The Cancer Genome Atlas) datasets. We further showed that the somatic mutations called by RNA-SSNV tended to have a higher functional impact and therapeutic power in known driver genes. Furthermore, VAF (variant allele fraction) analysis revealed that subclonal harboring expressed mutations had evolutional selection advantage and RNA had higher detection power to rescue DNA-omitted mutations. In sum, RNA-SSNV will be a useful approach to accurately call expressed somatic mutations for a more insightful analysis of cancer drive genes and carcinogenic mechanisms.
Keywords: RNA; RNA-SSNV; RNA-Seq; cancer; machine learning; somatic mutation.
Copyright © 2022 Long, Yuan and Li.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
RNAIndel: discovering somatic coding indels from tumor RNA-Seq data.Bioinformatics. 2020 Mar 1;36(5):1382-1390. doi: 10.1093/bioinformatics/btz753. Bioinformatics. 2020. PMID: 31593214 Free PMC article.
-
Detecting copy number alterations in RNA-Seq using SuperFreq.Bioinformatics. 2021 Nov 18;37(22):4023-4032. doi: 10.1093/bioinformatics/btab440. Bioinformatics. 2021. PMID: 34132781
-
FIREVAT: finding reliable variants without artifacts in human cancer samples using etiologically relevant mutational signatures.Genome Med. 2019 Dec 17;11(1):81. doi: 10.1186/s13073-019-0695-x. Genome Med. 2019. PMID: 31847917 Free PMC article.
-
Predicting Functional Effects of Synonymous Variants: A Systematic Review and Perspectives.Front Genet. 2019 Oct 7;10:914. doi: 10.3389/fgene.2019.00914. eCollection 2019. Front Genet. 2019. PMID: 31649718 Free PMC article.
-
vcfView: An Extensible Data Visualization and Quality Assurance Platform for Integrated Somatic Variant Analysis.Cancer Inform. 2020 Nov 11;19:1176935120972377. doi: 10.1177/1176935120972377. eCollection 2020. Cancer Inform. 2020. PMID: 33239857 Free PMC article. Review.
Cited by
-
Genotype prediction of 336,463 samples from public expression data.bioRxiv [Preprint]. 2024 Mar 13:2023.10.21.562237. doi: 10.1101/2023.10.21.562237. bioRxiv. 2024. PMID: 38559266 Free PMC article. Preprint.
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous