

Polymerases and DNA Repair in Neurons: Implications in Neuronal Survival and Neurodegenerative Diseases
- PMID: 35846567
- PMCID: PMC9279898
- DOI: 10.3389/fncel.2022.852002
Polymerases and DNA Repair in Neurons: Implications in Neuronal Survival and Neurodegenerative Diseases
Abstract
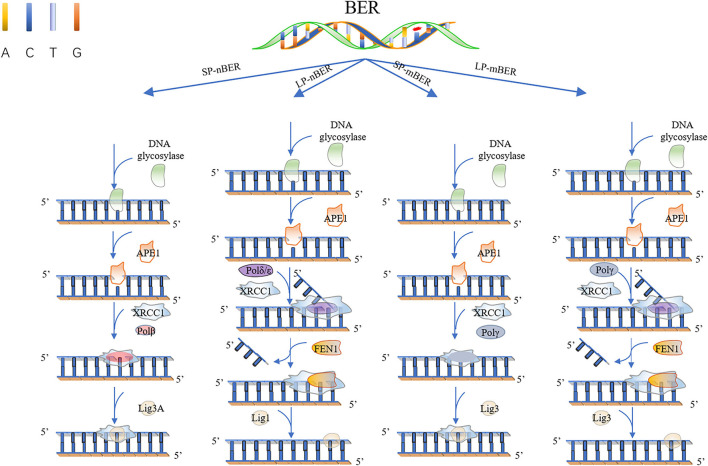
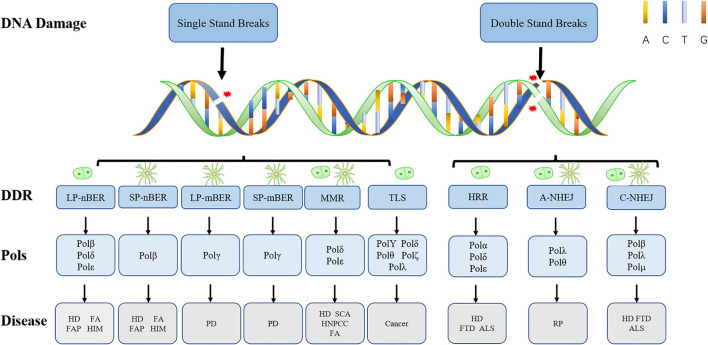
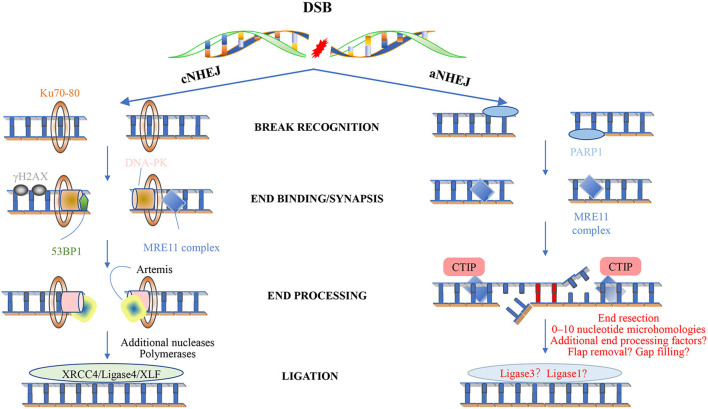
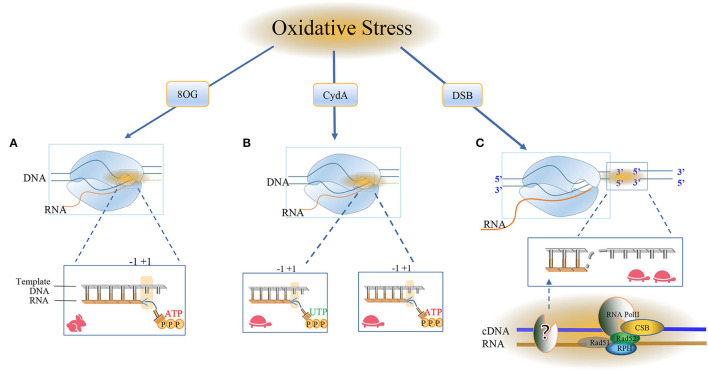
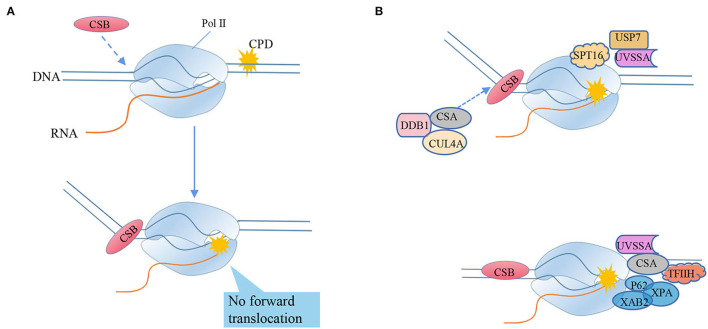
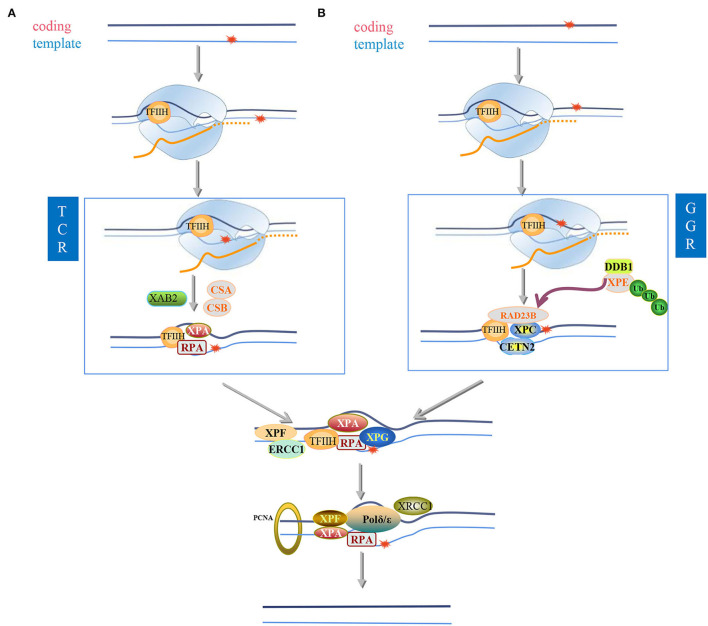
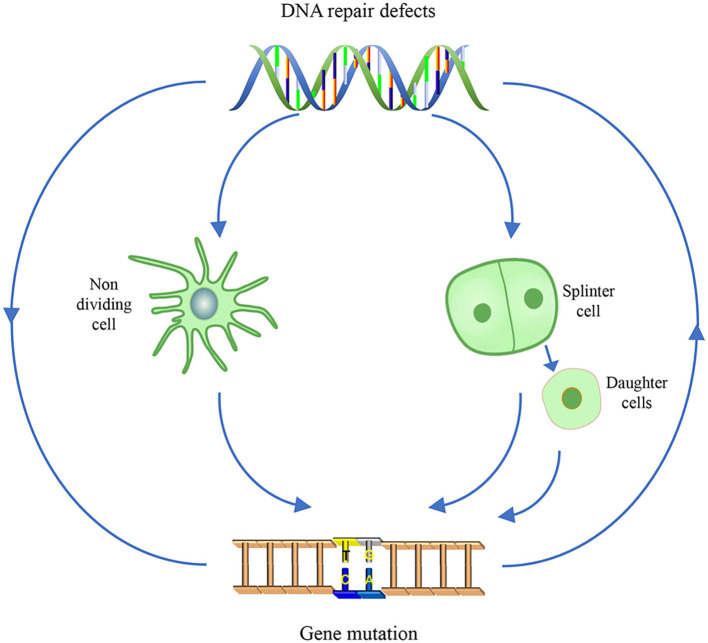
Most of the neurodegenerative diseases and aging are associated with reactive oxygen species (ROS) or other intracellular damaging agents that challenge the genome integrity of the neurons. As most of the mature neurons stay in G0/G1 phase, replication-uncoupled DNA repair pathways including BER, NER, SSBR, and NHEJ, are pivotal, efficient, and economic mechanisms to maintain genomic stability without reactivating cell cycle. In these progresses, polymerases are prominent, not only because they are responsible for both sensing and repairing damages, but also for their more diversified roles depending on the cell cycle phase and damage types. In this review, we summarized recent knowledge on the structural and biochemical properties of distinct polymerases, including DNA and RNA polymerases, which are known to be expressed and active in nervous system; the biological relevance of these polymerases and their interactors with neuronal degeneration would be most graphically illustrated by the neurological abnormalities observed in patients with hereditary diseases associated with defects in DNA repair; furthermore, the vicious cycle of the trinucleotide repeat (TNR) and impaired DNA repair pathway is also discussed. Unraveling the mechanisms and contextual basis of the role of the polymerases in DNA damage response and repair will promote our understanding about how long-lived postmitotic cells cope with DNA lesions, and why disrupted DNA repair contributes to disease origin, despite the diversity of mutations in genes. This knowledge may lead to new insight into the development of targeted intervention for neurodegenerative diseases.
Keywords: DNA polymerase; DNA repair pathway; RNA polymerase; neurodegenerative diseases; postmitotic cells.
Copyright © 2022 Li, Cao, Liu, Tang, Guo and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Almaguer-Mederos L. E., Jorge-Sainz Y., Almaguer-Gotay D., Aguilera-Rodríguez R., Rodríguez-Labrada R., Velázquez-Pérez L., et al. (2020). One-carbon metabolism factor MTHFR variant is associated with saccade latency in Spinocerebellar Ataxia type 2. J. Neurol. Sci. 409, 116586. 10.1016/j.jns.2019.116586 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials