

Characteristics and Genomic Diversity of Measles Virus From Measles Cases With Known Vaccination Status in Shanghai, China
- PMID: 35847814
- PMCID: PMC9281471
- DOI: 10.3389/fmed.2022.841650
Characteristics and Genomic Diversity of Measles Virus From Measles Cases With Known Vaccination Status in Shanghai, China
Abstract
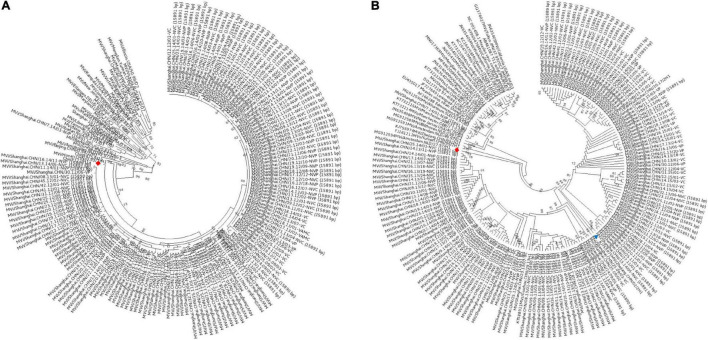
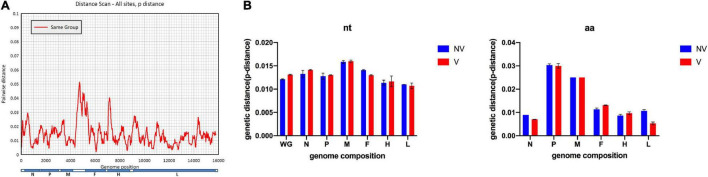
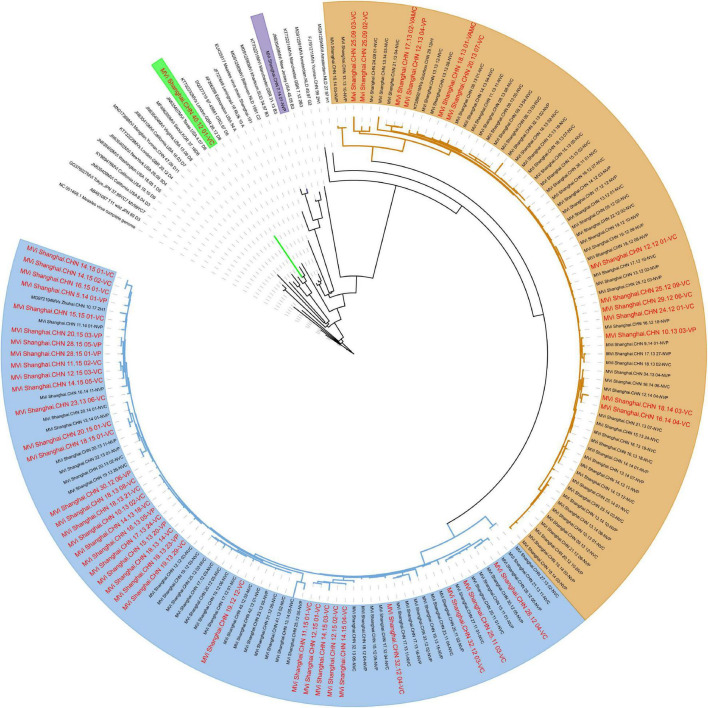
Although the highly effective measles vaccine has dramatically reduced the incidence of measles, measles, and outbreaks continue to occur in individuals who received the measles vaccine because of immunization failure. In this study, patients who have definite records of immunization were enrolled based on measles surveillance in Shanghai, China, from 2009 to 2017, and genomic characteristics regarding viruses retrieved from these cases provided insights into immunization failure. A total of 147 complete genomes of measles virus (MV) were obtained from the laboratory-confirmed cases through Illumina MiSeq. Epidemiological, and genetic characteristics of the MV were focused on information about age, gender, immunization record, variation, and evolution of the whole genome. Furthermore, systematic genomics using phylogeny and selection pressure approaches were analyzed. Our analysis based on the whole genome of 147 isolates revealed 4 clusters: 2 for the genotype H1 (clusters named H1-A, including 73 isolates; H1-B, including 72 isolates) and the other 2 for D8 and B3, respectively. Estimated nucleotide substitution rates of genotype H1 MV derived using genome and individual genes are lower than other genotypes. Our study contributes to global measles epidemiology and proves that whole-genome sequencing was a useful tool for more refined genomic characterization. The conclusion indicates that vaccination may have an effect on virus evolution. However, no major impact was found on the antigenicity in Shanghai isolates.
Keywords: Shanghai isolates; comparative genomics; complete genome sequences; genetic diversity; measles virus; vaccine failure.
Copyright © 2022 Cui, Li, Yang, Tang, Li, Chen, Li, Cui, Huang, Sun, Xu, Zhang, Li and Zhang.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Characterization of diversity of measles viruses in India: Genomic sequencing and comparative genomics studies.J Infect. 2020 Mar;80(3):301-309. doi: 10.1016/j.jinf.2019.11.025. Epub 2020 Jan 17. J Infect. 2020. PMID: 31958542
-
Molecular characterization of measles viruses in China: Circulation dynamics of the endemic H1 genotype from 2011 to 2017.PLoS One. 2019 Jun 20;14(6):e0218782. doi: 10.1371/journal.pone.0218782. eCollection 2019. PLoS One. 2019. PMID: 31220172 Free PMC article.
-
Molecular evolution and genomic characteristics of genotype H1 of measles virus.J Med Virol. 2022 Feb;94(2):521-530. doi: 10.1002/jmv.27448. Epub 2021 Nov 18. J Med Virol. 2022. PMID: 34761827
-
[Genetic characterization of the isolates of measles viruses in Hongkou district of Shanghai, China in 2012].Zhonghua Liu Xing Bing Xue Za Zhi. 2014 Apr;35(4):429-32. Zhonghua Liu Xing Bing Xue Za Zhi. 2014. PMID: 25009035 Chinese.
-
Epidemiological and genetic characterization of measles virus circulating strains at Marseille, France during 2017-2019 measles outbreak.J Infect. 2021 Sep;83(3):361-370. doi: 10.1016/j.jinf.2021.07.011. Epub 2021 Jul 24. J Infect. 2021. PMID: 34310945
Cited by
-
Genomic tools for post-elimination measles molecular epidemiology using Canadian surveillance data from 2018-2020.Front Microbiol. 2024 Nov 19;15:1475144. doi: 10.3389/fmicb.2024.1475144. eCollection 2024. Front Microbiol. 2024. PMID: 39629208 Free PMC article.
References
-
- Griffin DE. Measles virus. Fields Virol. (2013):1042–8.
-
- World Health Organization. World health organization measles virus nomenclature update. Wkly Epidemiol Rec. (2012) 87:73–81.
-
- No authors. The role of extended and whole genome sequencing for tracking transmission of measles and rubella viruses: report from the Global Measles and Rubella Laboratory Network meeting, 2017. Wkly Epidemiol Rec. (2018) 93:55–9. - PubMed
LinkOut - more resources
Full Text Sources