

Protein Structural Modeling for Electron Microscopy Maps Using VESPER and MAINMAST
- PMID: 35849043
- PMCID: PMC9299282
- DOI: 10.1002/cpz1.494
Protein Structural Modeling for Electron Microscopy Maps Using VESPER and MAINMAST
Abstract
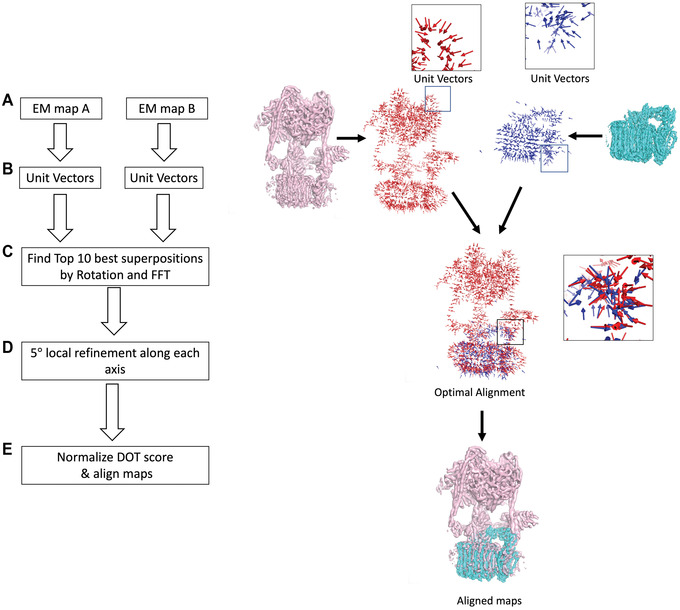
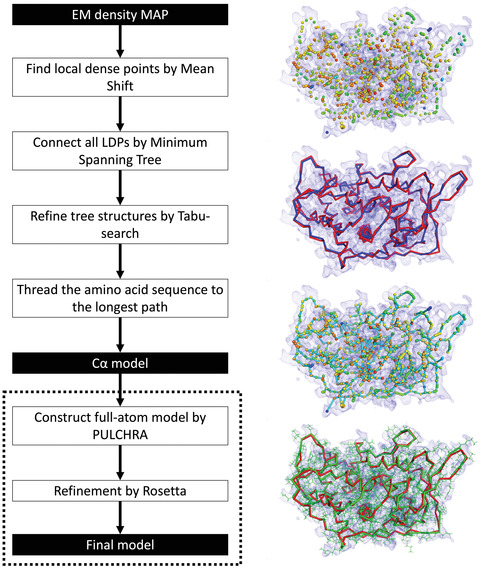
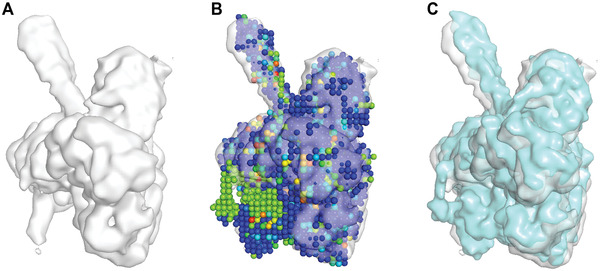
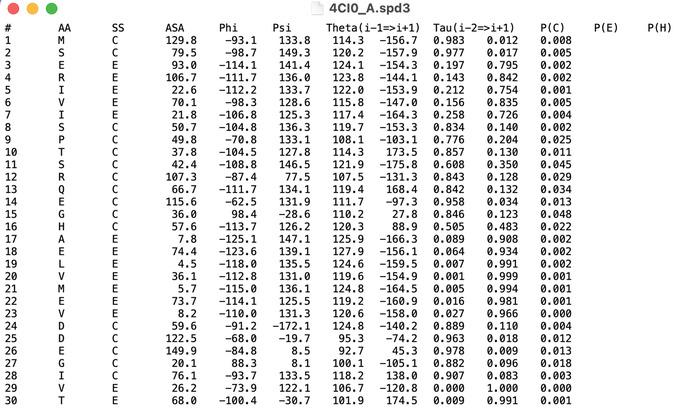
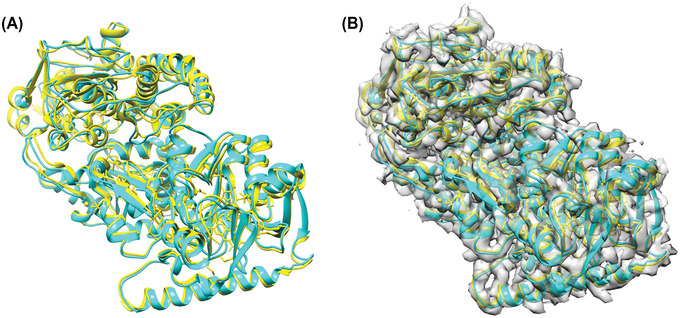
An increasing number of protein structures are determined by cryo-electron microscopy (cryo-EM) and stored in the Electron Microscopy Data Bank (EMDB). To interpret determined cryo-EM maps, several methods have been developed that model the tertiary structure of biomolecules, particularly proteins. Here we show how to use two such methods, VESPER and MAINMAST, which were developed in our group. VESPER is a method mainly for two purposes: fitting protein structure models into an EM map and aligning two EM maps locally or globally to capture their similarity. VESPER represents each EM map as a set of vectors pointing toward denser points. By considering matching the directions of vectors, in general, VESPER aligns maps better than conventional methods that only consider local densities of maps. MAINMAST is a de novo protein modeling tool designed for EM maps with resolution of 3-5 Å or better. MAINMAST builds a protein main chain directly from a density map by tracing dense points in an EM map and connecting them using a tree-graph structure. This article describes how to use these two tools using three illustrative modeling examples. © 2022 The Authors. Current Protocols published by Wiley Periodicals LLC. Basic Protocol 1: Protein structure model fitting using VESPER Alternate Protocol: Atomic model fitting using VESPER web server Basic Protocol 2: Protein de novo modeling using MAINMAST.
Keywords: cryo-EM alignment; de novo protein modeling; electron microscopy maps; protein fitting; protein structure.
© 2022 The Authors. Current Protocols published by Wiley Periodicals LLC.
Conflict of interest statement
The authors declare no competing interests.
Figures

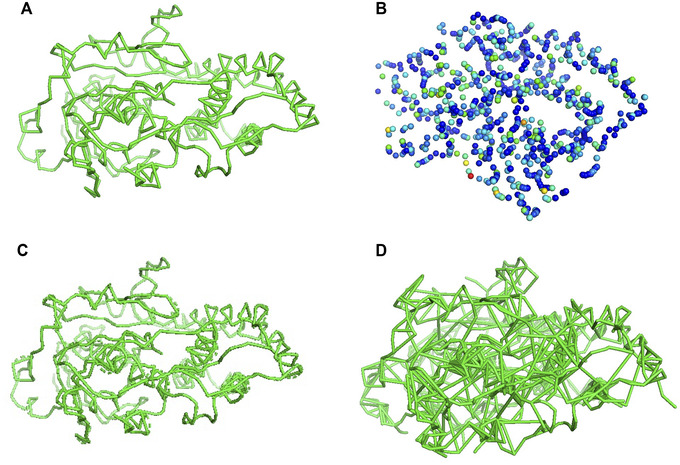
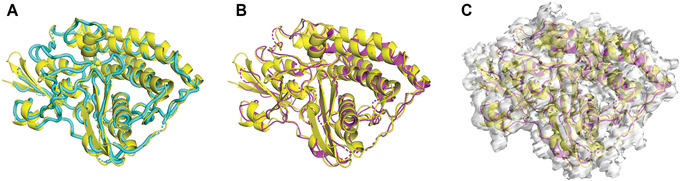
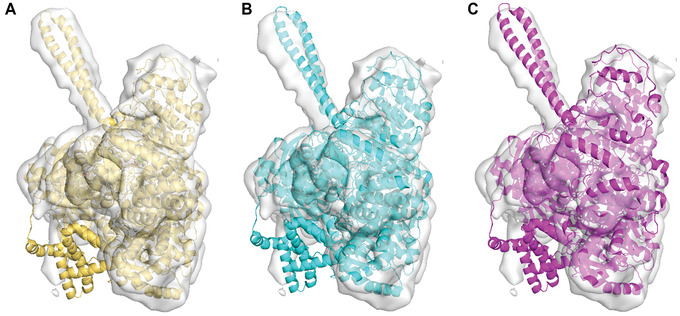
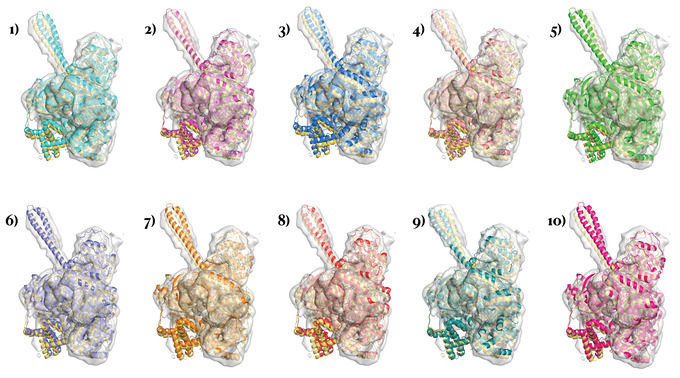
Similar articles
-
Protein Structure Modeling from Cryo-EM Map Using MAINMAST and MAINMAST-GUI Plugin.Methods Mol Biol. 2020;2165:317-336. doi: 10.1007/978-1-0716-0708-4_19. Methods Mol Biol. 2020. PMID: 32621234
-
VESPER: global and local cryo-EM map alignment using local density vectors.Nat Commun. 2021 Apr 7;12(1):2090. doi: 10.1038/s41467-021-22401-y. Nat Commun. 2021. PMID: 33828103 Free PMC article.
-
De novo main-chain modeling for EM maps using MAINMAST.Nat Commun. 2018 Apr 24;9(1):1618. doi: 10.1038/s41467-018-04053-7. Nat Commun. 2018. PMID: 29691408 Free PMC article.
-
Advancing structure modeling from cryo-EM maps with deep learning.Biochem Soc Trans. 2025 Feb 7;53(1):259-65. doi: 10.1042/BST20240784. Biochem Soc Trans. 2025. PMID: 39927816 Free PMC article. Review.
-
Evolution of standardization and dissemination of cryo-EM structures and data jointly by the community, PDB, and EMDB.J Biol Chem. 2021 Jan-Jun;296:100560. doi: 10.1016/j.jbc.2021.100560. Epub 2021 Mar 18. J Biol Chem. 2021. PMID: 33744287 Free PMC article. Review.
Cited by
-
Cryo2StructData: A Large Labeled Cryo-EM Density Map Dataset for AI-based Modeling of Protein Structures.bioRxiv [Preprint]. 2024 Jan 2:2023.06.14.545024. doi: 10.1101/2023.06.14.545024. bioRxiv. 2024. Update in: Sci Data. 2024 May 6;11(1):458. doi: 10.1038/s41597-024-03299-9. PMID: 37398020 Free PMC article. Updated. Preprint.
-
Cryo2StructData: A Large Labeled Cryo-EM Density Map Dataset for AI-based Modeling of Protein Structures.Sci Data. 2024 May 6;11(1):458. doi: 10.1038/s41597-024-03299-9. Sci Data. 2024. PMID: 38710720 Free PMC article.
-
Deep learning for reconstructing protein structures from cryo-EM density maps: Recent advances and future directions.Curr Opin Struct Biol. 2023 Apr;79:102536. doi: 10.1016/j.sbi.2023.102536. Epub 2023 Feb 9. Curr Opin Struct Biol. 2023. PMID: 36773336 Free PMC article. Review.
-
Protein Secondary Structure and DNA/RNA Detection for Cryo-EM and Cryo-ET Using Emap2sec and Emap2sec.Methods Mol Biol. 2025;2867:105-120. doi: 10.1007/978-1-0716-4196-5_6. Methods Mol Biol. 2025. PMID: 39576577
References
-
- Carreira‐Perpinan, M. A. (2006). Acceleration strategies for gaussian mean‐shift image segmentation. IEEE Conference on Computer Vision and Pattern Recognition, 1, 1160–1167.
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
