

Recommendations for clinical interpretation of variants found in non-coding regions of the genome
- PMID: 35850704
- PMCID: PMC9295495
- DOI: 10.1186/s13073-022-01073-3
Recommendations for clinical interpretation of variants found in non-coding regions of the genome
Abstract
Background: The majority of clinical genetic testing focuses almost exclusively on regions of the genome that directly encode proteins. The important role of variants in non-coding regions in penetrant disease is, however, increasingly being demonstrated, and the use of whole genome sequencing in clinical diagnostic settings is rising across a large range of genetic disorders. Despite this, there is no existing guidance on how current guidelines designed primarily for variants in protein-coding regions should be adapted for variants identified in other genomic contexts.
Methods: We convened a panel of nine clinical and research scientists with wide-ranging expertise in clinical variant interpretation, with specific experience in variants within non-coding regions. This panel discussed and refined an initial draft of the guidelines which were then extensively tested and reviewed by external groups.
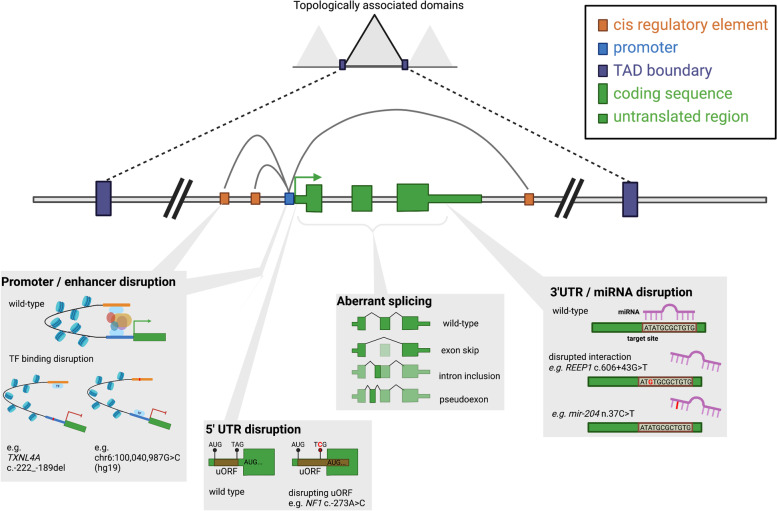
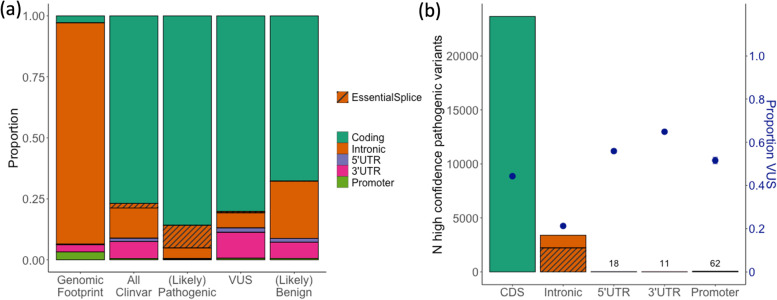
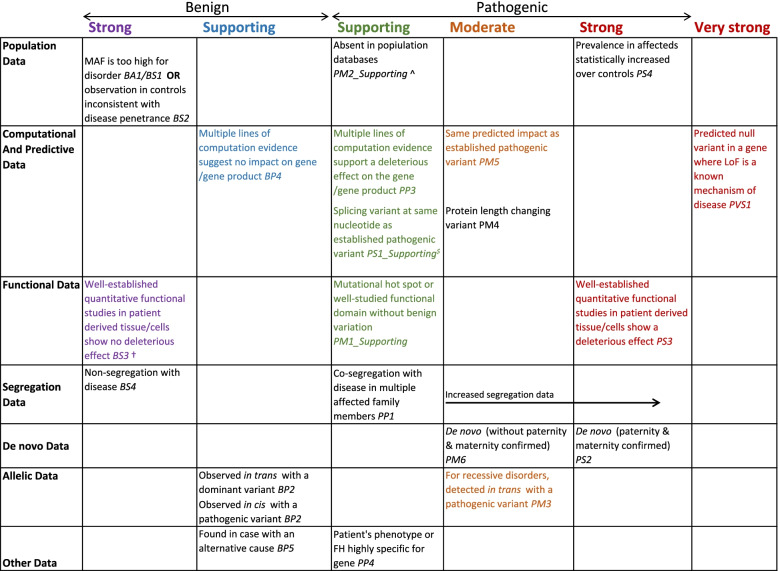
Results: We discuss considerations specifically for variants in non-coding regions of the genome. We outline how to define candidate regulatory elements, highlight examples of mechanisms through which non-coding region variants can lead to penetrant monogenic disease, and outline how existing guidelines can be adapted for the interpretation of these variants.
Conclusions: These recommendations aim to increase the number and range of non-coding region variants that can be clinically interpreted, which, together with a compatible phenotype, can lead to new diagnoses and catalyse the discovery of novel disease mechanisms.
Keywords: Gene regulation; Non-coding variation; Variant interpretation.
© 2022. The Author(s).
Conflict of interest statement
AODL is a paid member of the Scientific Advisory Board of Congenica. SMH is a paid employee of Ambry Genetics. All other authors declare that they have no competing interests.
Figures
References
-
- Caulfield M, Davies J, Dennys M, Elbahy L, Fowler T, Hill S, et al. The national genomics research and healthcare knowledgebase. figshare; 2019.
-
- Ellingford JM, Barton S, Bhaskar S, Williams SG, Sergouniotis PI, O’Sullivan J, et al. Whole genome sequencing increases molecular diagnostic yield compared with current diagnostic testing for inherited retinal disease. Ophthalmology. 2016;123:1143–1150. doi: 10.1016/j.ophtha.2016.01.009. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
