

Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC
- PMID: 35851854
- PMCID: PMC9352586
- DOI: 10.1038/s41564-022-01185-x
Early detection and surveillance of SARS-CoV-2 genomic variants in wastewater using COJAC
Abstract
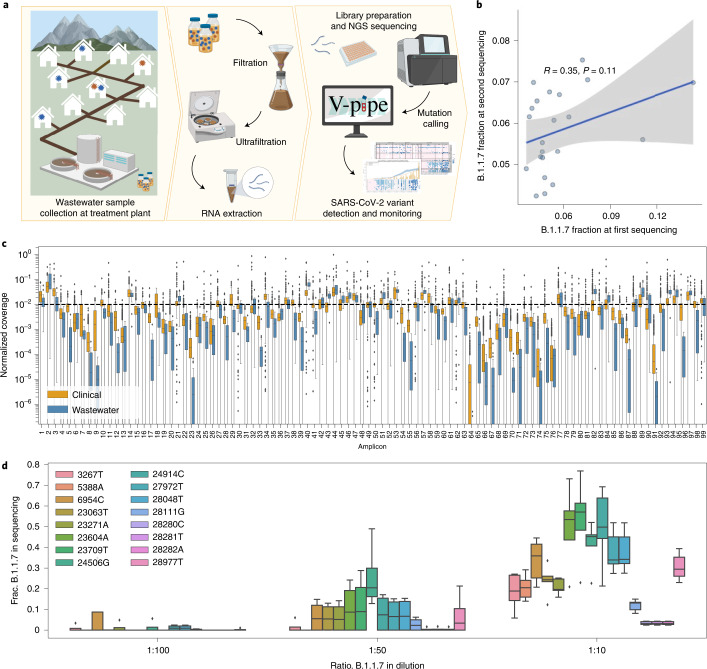
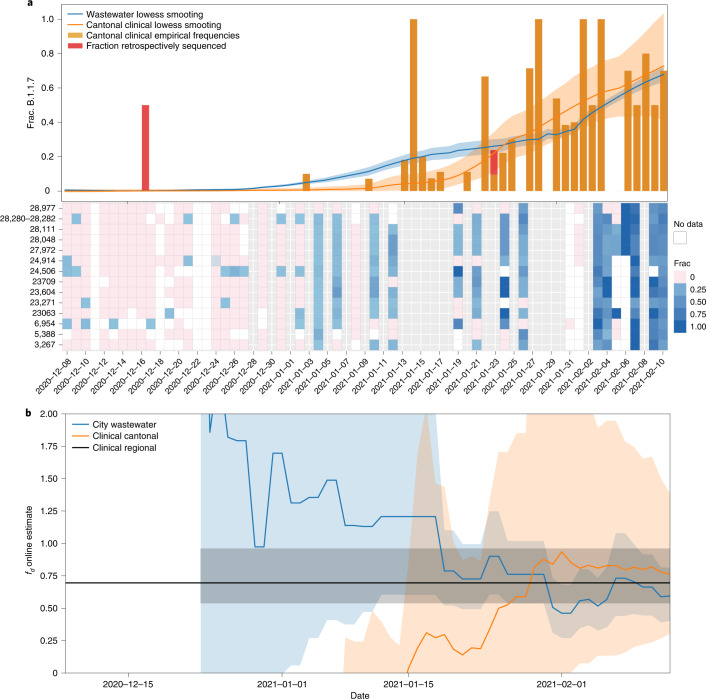
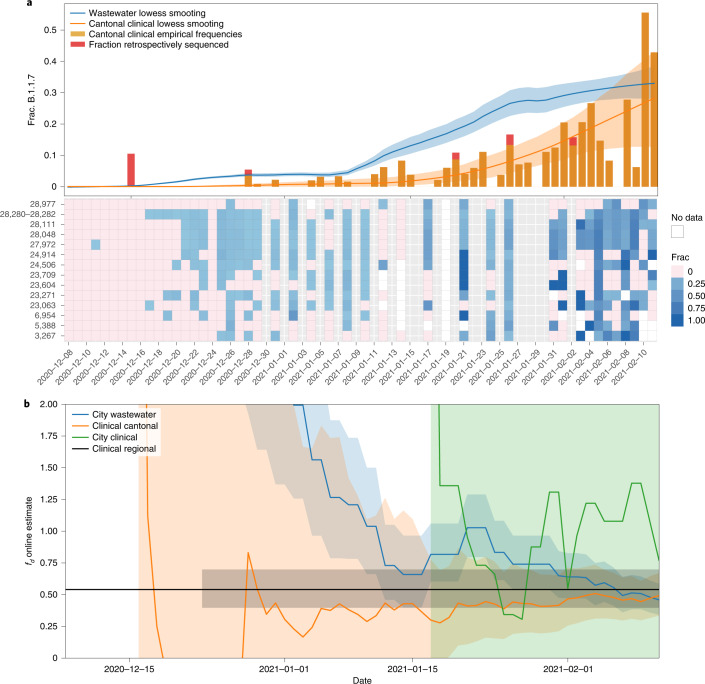
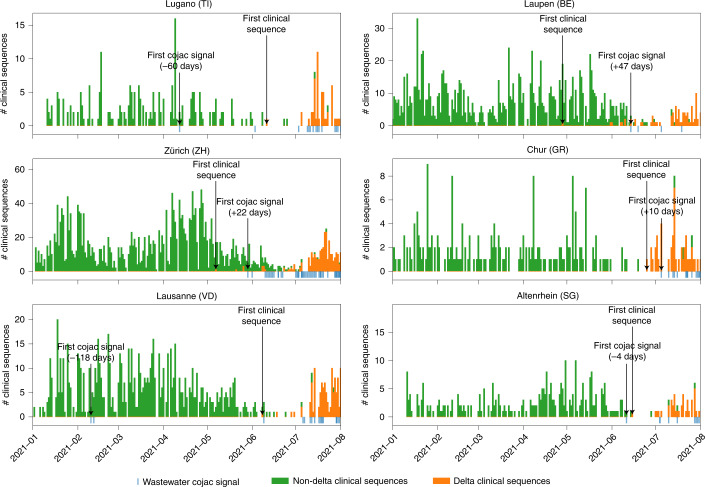
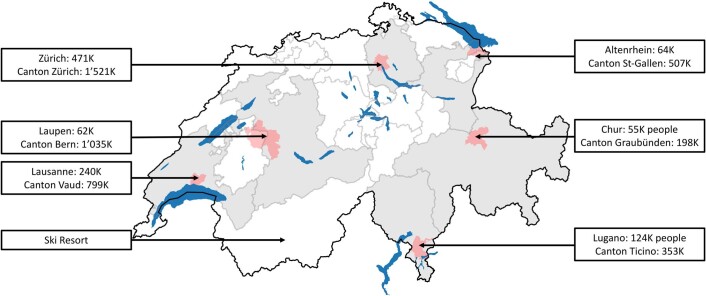
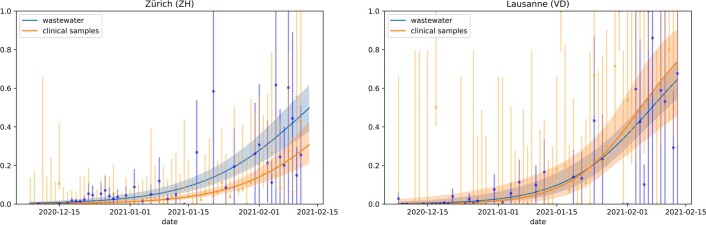
The continuing emergence of SARS-CoV-2 variants of concern and variants of interest emphasizes the need for early detection and epidemiological surveillance of novel variants. We used genomic sequencing of 122 wastewater samples from three locations in Switzerland to monitor the local spread of B.1.1.7 (Alpha), B.1.351 (Beta) and P.1 (Gamma) variants of SARS-CoV-2 at a population level. We devised a bioinformatics method named COJAC (Co-Occurrence adJusted Analysis and Calling) that uses read pairs carrying multiple variant-specific signature mutations as a robust indicator of low-frequency variants. Application of COJAC revealed that a local outbreak of the Alpha variant in two Swiss cities was observable in wastewater up to 13 d before being first reported in clinical samples. We further confirmed the ability of COJAC to detect emerging variants early for the Delta variant by analysing an additional 1,339 wastewater samples. While sequencing data of single wastewater samples provide limited precision for the quantification of relative prevalence of a variant, we show that replicate and close-meshed longitudinal sequencing allow for robust estimation not only of the local prevalence but also of the transmission fitness advantage of any variant. We conclude that genomic sequencing and our computational analysis can provide population-level estimates of prevalence and fitness of emerging variants from wastewater samples earlier and on the basis of substantially fewer samples than from clinical samples. Our framework is being routinely used in large national projects in Switzerland and the UK.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
-
- Tegally, H. et al. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. Preprint at medRxiv10.1101/2020.12.21.20248640 (2020).
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
