

Upfront Biology-Guided Therapy in Diffuse Intrinsic Pontine Glioma: Therapeutic, Molecular, and Biomarker Outcomes from PNOC003
- PMID: 35852795
- PMCID: PMC9475246
- DOI: 10.1158/1078-0432.CCR-22-0803
Upfront Biology-Guided Therapy in Diffuse Intrinsic Pontine Glioma: Therapeutic, Molecular, and Biomarker Outcomes from PNOC003
Abstract
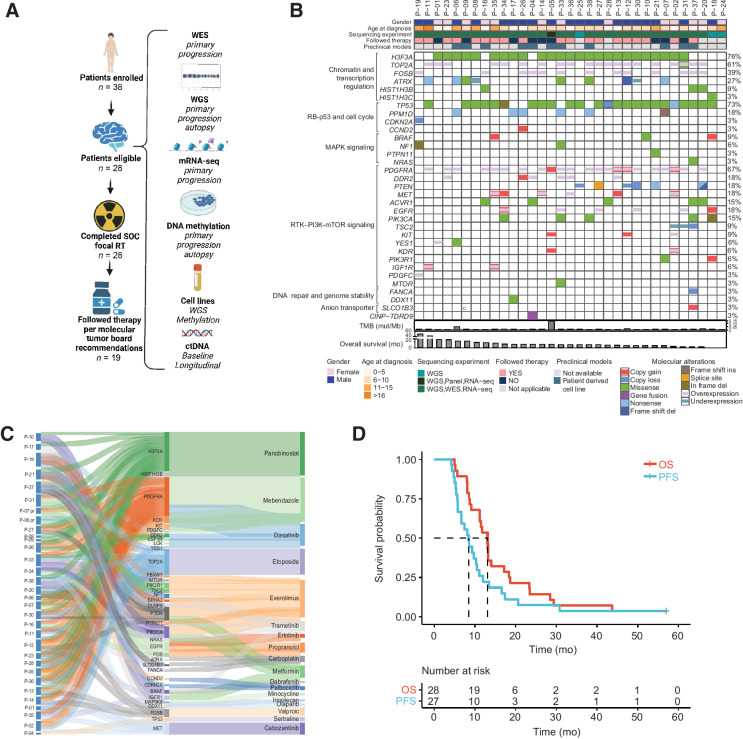
Purpose: PNOC003 is a multicenter precision medicine trial for children and young adults with newly diagnosed diffuse intrinsic pontine glioma (DIPG).
Patients and methods: Patients (3-25 years) were enrolled on the basis of imaging consistent with DIPG. Biopsy tissue was collected for whole-exome and mRNA sequencing. After radiotherapy (RT), patients were assigned up to four FDA-approved drugs based on molecular tumor board recommendations. H3K27M-mutant circulating tumor DNA (ctDNA) was longitudinally measured. Tumor tissue and matched primary cell lines were characterized using whole-genome sequencing and DNA methylation profiling. When applicable, results were verified in an independent cohort from the Children's Brain Tumor Network (CBTN).
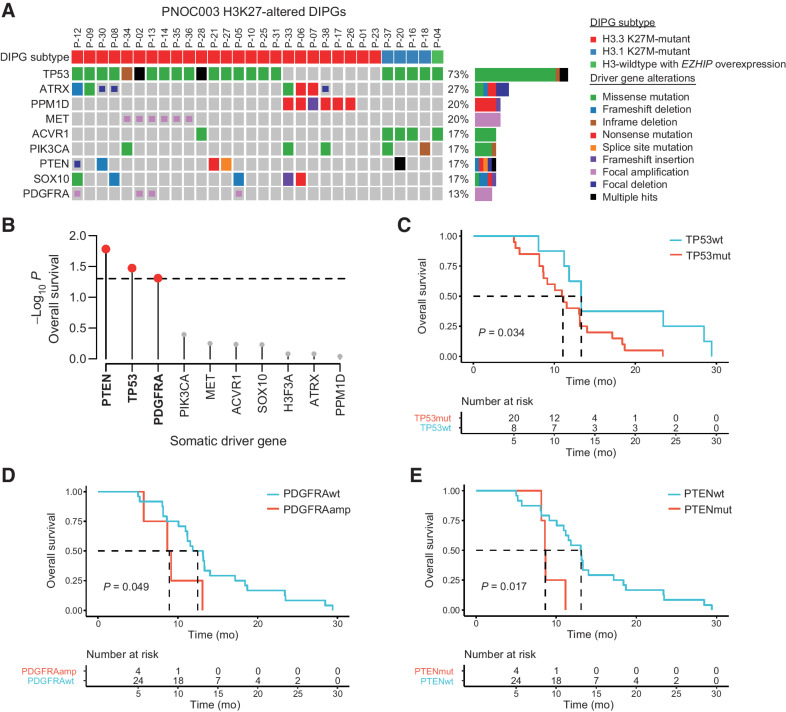
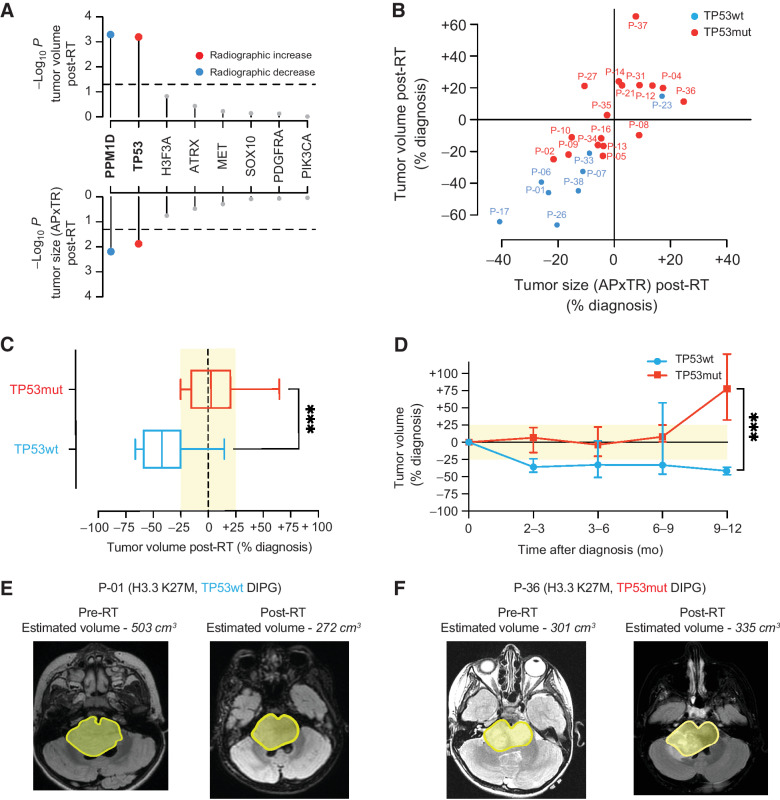
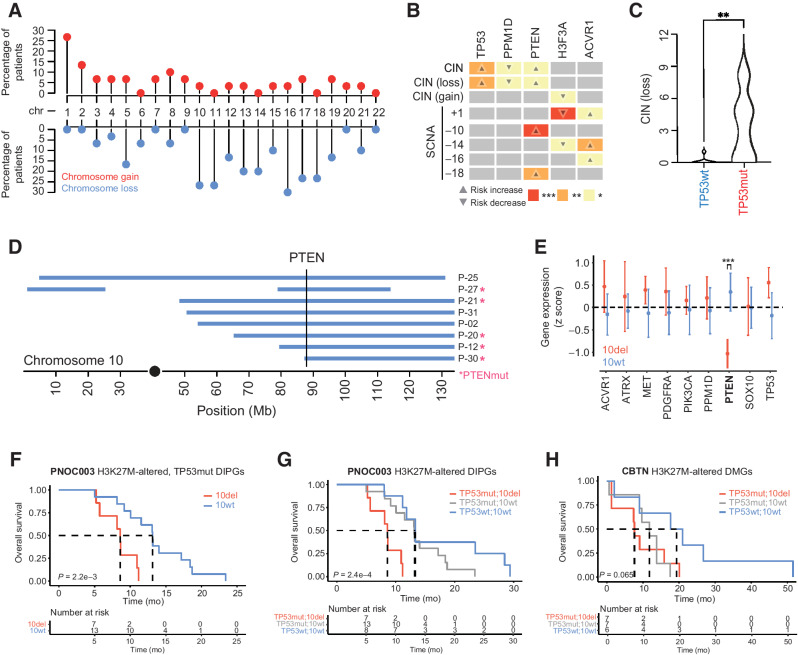
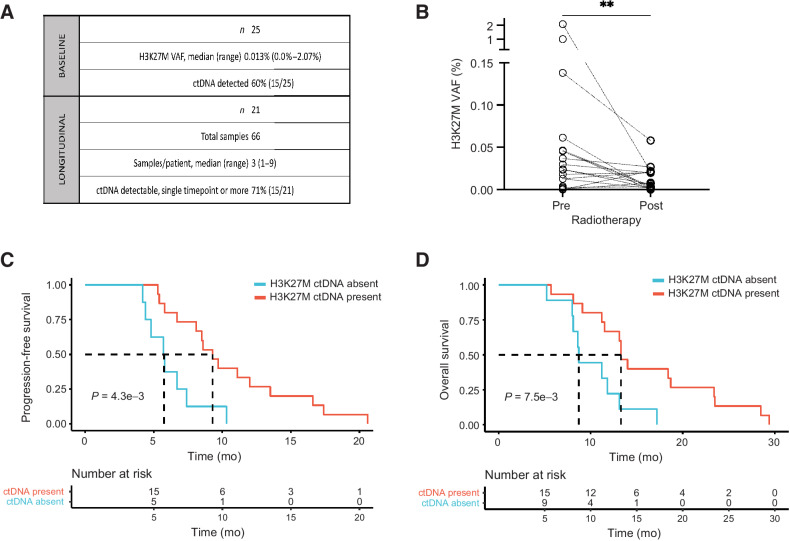
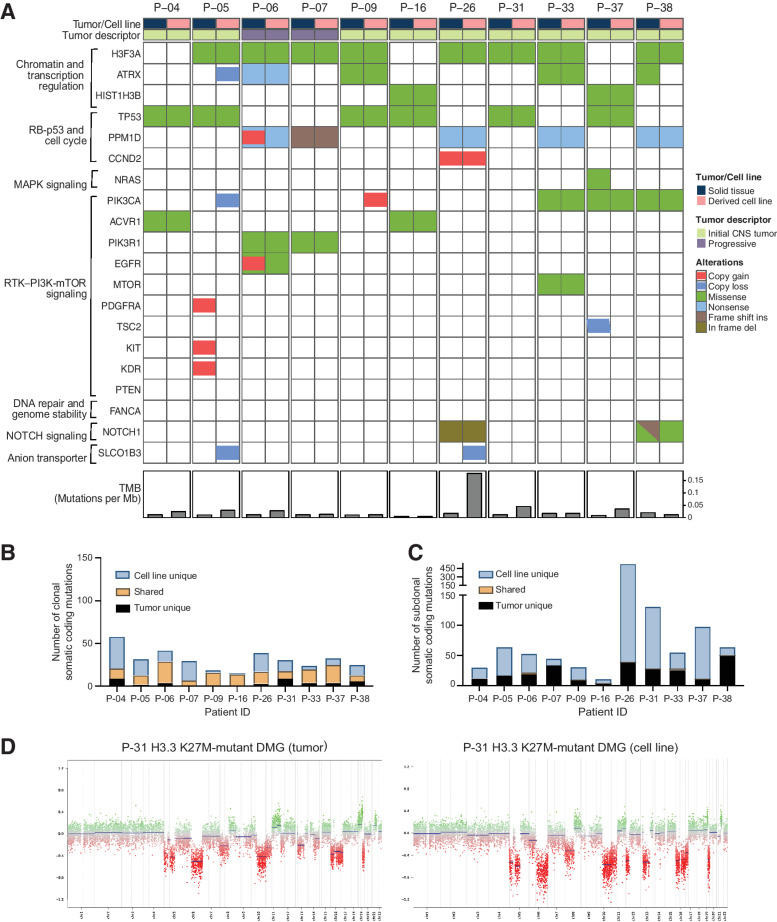
Results: Of 38 patients enrolled, 28 patients (median 6 years, 10 females) were reviewed by the molecular tumor board. Of those, 19 followed treatment recommendations. Median overall survival (OS) was 13.1 months [95% confidence interval (CI), 11.2-18.4] with no difference between patients who followed recommendations and those who did not. H3K27M-mutant ctDNA was detected at baseline in 60% of cases tested and associated with response to RT and survival. Eleven cell lines were established, showing 100% fidelity of key somatic driver gene alterations in the primary tumor. In H3K27-altered DIPGs, TP53 mutations were associated with worse OS (TP53mut 11.1 mo; 95% CI, 8.7-14; TP53wt 13.3 mo; 95% CI, 11.8-NA; P = 3.4e-2), genome instability (P = 3.1e-3), and RT resistance (P = 6.4e-4). The CBTN cohort confirmed an association between TP53 mutation status, genome instability, and clinical outcome.
Conclusions: Upfront treatment-naïve biopsy provides insight into clinically relevant molecular alterations and prognostic biomarkers for H3K27-altered DIPGs.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Macy ME, Kieran MW, Chi SN, Cohen KJ, MacDonald TJ, Smith AA, et al. A pediatric trial of radiation/cetuximab followed by irinotecan/cetuximab in newly diagnosed diffuse pontine gliomas and high-grade astrocytomas: A Pediatric Oncology Experimental Therapeutics Investigators' Consortium study. Pediatr Blood Cancer. 2017 Nov;64(11):10.1002/pbc.26621. doi: 10.1002/pbc.26621. Epub 2017 May 24. PMID: 28544128; PMCID: PMC5605460. - PMC - PubMed
-
- Veldhuijzen van Zanten SE, Jansen MH, Sanchez Aliaga E, van Vuurden DG, Vandertop WP, Kaspers GJ. A twenty-year review of diagnosing and treating children with diffuse intrinsic pontine glioma in the Netherlands. Expert Rev Anticancer Ther 2015;15:157–64. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
