

HAP40 is a conserved central regulator of Huntingtin and a potential modulator of Huntington's disease pathogenesis
- PMID: 35853002
- PMCID: PMC9295956
- DOI: 10.1371/journal.pgen.1010302
HAP40 is a conserved central regulator of Huntingtin and a potential modulator of Huntington's disease pathogenesis
Abstract
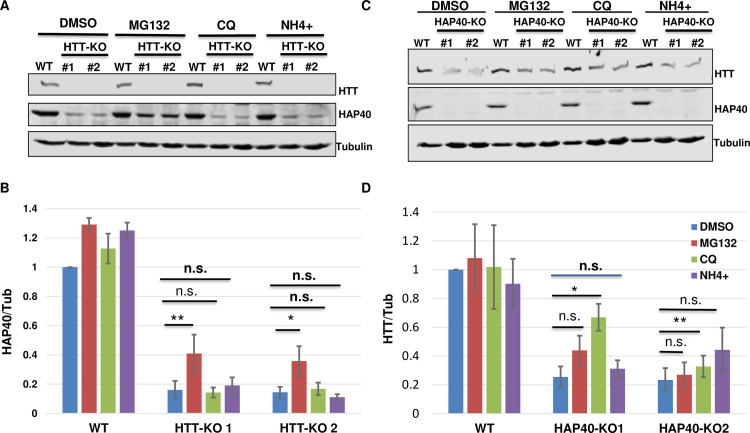
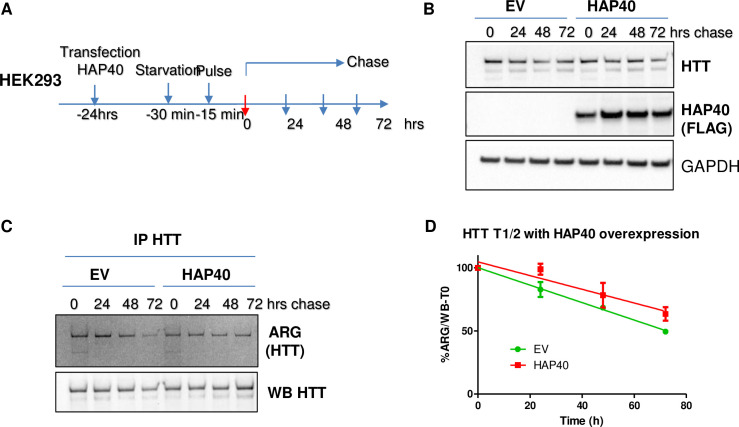
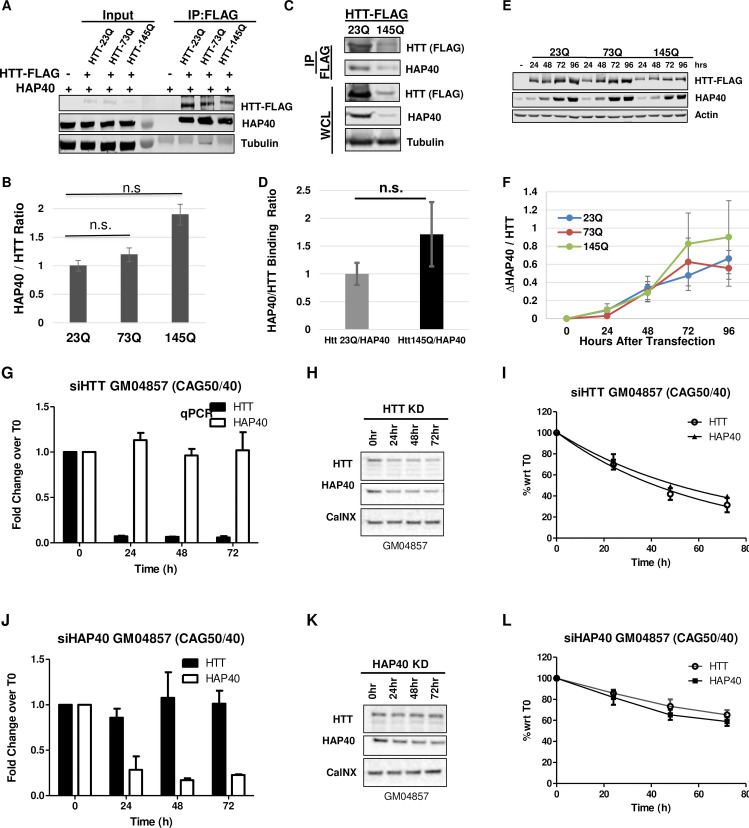
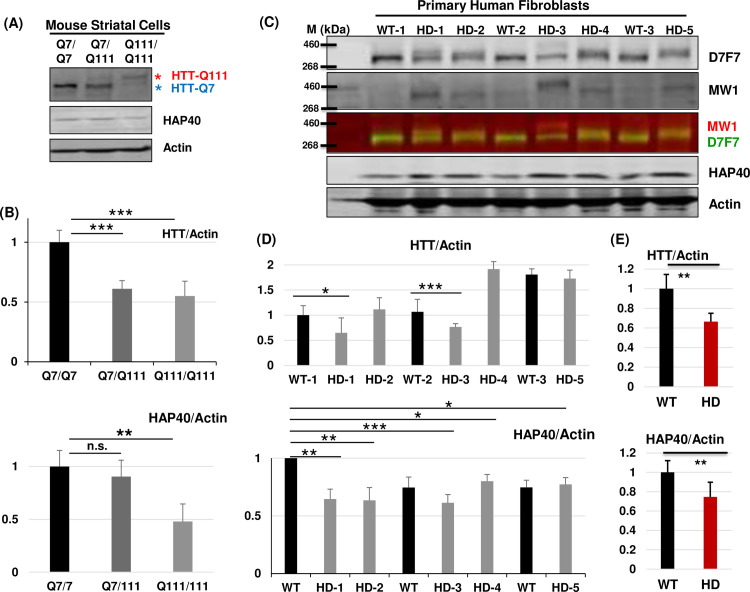
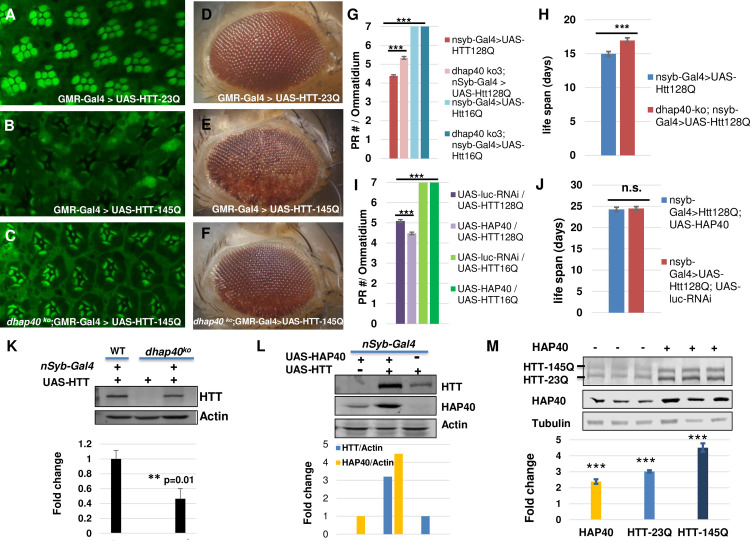
Perturbation of huntingtin (HTT)'s physiological function is one postulated pathogenic factor in Huntington's disease (HD). However, little is known how HTT is regulated in vivo. In a proteomic study, we isolated a novel ~40kDa protein as a strong binding partner of Drosophila HTT and demonstrated it was the functional ortholog of HAP40, an HTT associated protein shown recently to modulate HTT's conformation but with unclear physiological and pathologic roles. We showed that in both flies and human cells, HAP40 maintained conserved physical and functional interactions with HTT. Additionally, loss of HAP40 resulted in similar phenotypes as HTT knockout. More strikingly, HAP40 strongly affected HTT's stability, as depletion of HAP40 significantly reduced the levels of endogenous HTT protein while HAP40 overexpression markedly extended its half-life. Conversely, in the absence of HTT, the majority of HAP40 protein were degraded, likely through the proteasome. Further, the affinity between HTT and HAP40 was not significantly affected by polyglutamine expansion in HTT, and contrary to an early report, there were no abnormal accumulations of endogenous HAP40 protein in HD cells from mouse HD models or human patients. Lastly, when tested in Drosophila models of HD, HAP40 partially modulated the neurodegeneration induced by full-length mutant HTT while showed no apparent effect on the toxicity of mutant HTT exon 1 fragment. Together, our study uncovers a conserved mechanism governing the stability and in vivo functions of HTT and demonstrates that HAP40 is a central and positive regulator of endogenous HTT. Further, our results support that mutant HTT is toxic regardless of the presence of its partner HAP40, and implicate HAP40 as a potential modulator of HD pathogenesis through its multiplex effect on HTT's function, stability and the potency of mutant HTT's toxicity.
Conflict of interest statement
I have read the journal’s policy and the authors of this manuscript have the following competing interests: GM and DM are employed by CHDI Management, Inc. as advisors to CHDI Foundation, Inc., and declare no conflict of interest. CHDI Foundation is a nonprofit biomedical research organization exclusively dedicated to collaboratively developing therapeutics that substantially improve the lives of those affected by Huntington’s disease. CHDI Foundation conducts research in a number of different ways; for the purposes of this manuscript, all research was conceptualized, planned, and directed by all authors and conducted at the University of Texas Health Science Center at Houston (UTHealth) or the CHDI-supported contract research organization IRBM. This does not alter our adherence to PLOS Genetics policies on sharing data and materials.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
