

A recurrent single-amino acid deletion (p.Glu500del) in the head domain of ß-cardiac myosin in two unrelated boys presenting with polyhydramnios, congenital axial stiffness and skeletal myopathy
- PMID: 35854315
- PMCID: PMC9295345
- DOI: 10.1186/s13023-022-02421-7
A recurrent single-amino acid deletion (p.Glu500del) in the head domain of ß-cardiac myosin in two unrelated boys presenting with polyhydramnios, congenital axial stiffness and skeletal myopathy
Abstract
Background: Alterations in the MYH7 gene can cause cardiac and skeletal myopathies. MYH7-related skeletal myopathies are extremely rare, and the vast majority of causal variants in the MYH7 gene are predicted to alter the rod domain of the of ß-cardiac myosin molecule, resulting in distal muscle weakness as the predominant manifestation. Here we describe two unrelated patients harboring an in-frame deletion in the MYH7 gene that is predicted to result in deletion of a single amino acid (p.Glu500del) in the head domain of ß-cardiac myosin. Both patients display an unusual skeletal myopathy phenotype with congenital axial stiffness and muscular hypertonus, but no cardiac involvement.
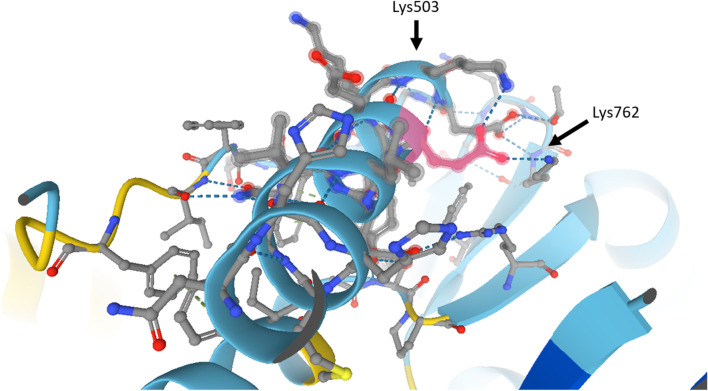
Results: Clinical data, MRI results and histopathological data were collected retrospectively in two unrelated boys (9 and 3.5 years old). Exome sequencing uncovered the same 3-bp in-frame deletion in exon 15 (c.1498_1500delGAG) of the MYH7 gene of both patients, a mutation which deletes a highly conserved glutamate residue (p.Glu500del) in the relay loop of the head domain of the ß-cardiac myosin heavy chain. The mutation occurred de novo in one patient, whereas mosaicism was detected in blood of the father of the second patient. Both boys presented with an unusual phenotype of prenatal polyhydramnios, congenital axial stiffness and muscular hypertonus. In one patient the phenotype evolved into an axial/proximal skeletal myopathy without distal involvement or cardiomyopathy, whereas the other patient exhibited predominantly stiffness and respiratory involvement. We review and compare all patients described in the literature who possess a variant predicted to alter the p.Glu500 residue in the ß-cardiac myosin head domain, and we provide in-silico analyses of potential effects on polypeptide function.
Conclusion: The data presented here expand the phenotypic spectrum of mutations in the MYH7 gene and have implications for future diagnostics and therapeutic approaches.
Keywords: Actin-binding domain; Exome-analysis; MYH7; Myopathy; ß-cardiac myosin heavy chain.
© 2022. The Author(s).
Conflict of interest statement
The authors declare they have no competing interests.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
