

Genetic etiology and clinical challenges of phenylketonuria
- PMID: 35854334
- PMCID: PMC9295449
- DOI: 10.1186/s40246-022-00398-9
Genetic etiology and clinical challenges of phenylketonuria
Abstract
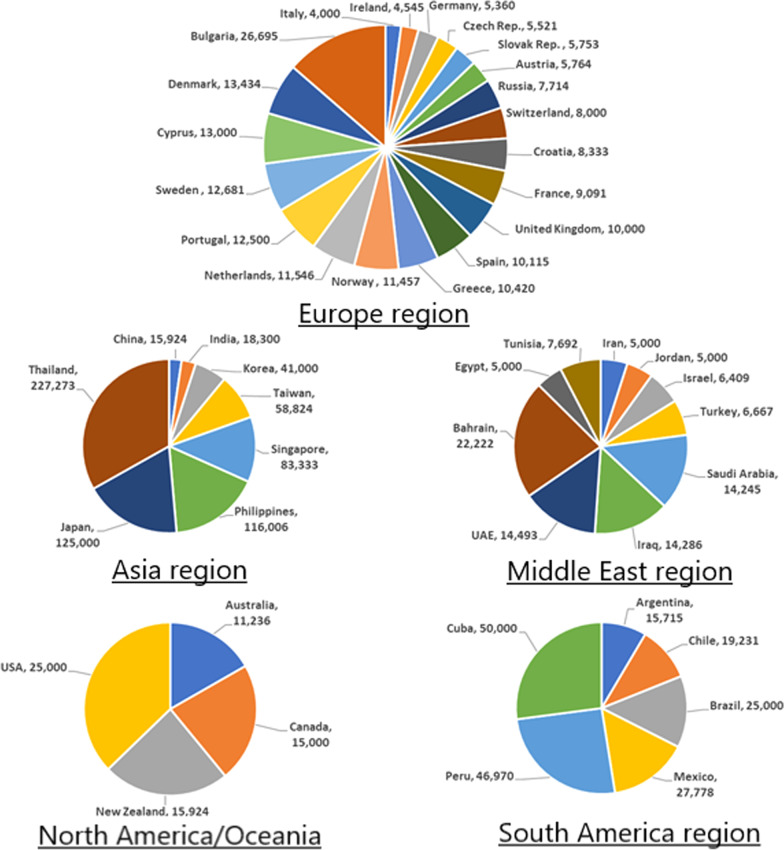
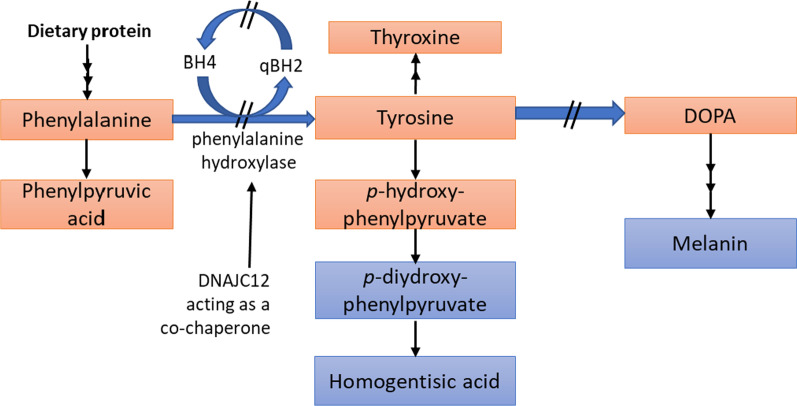
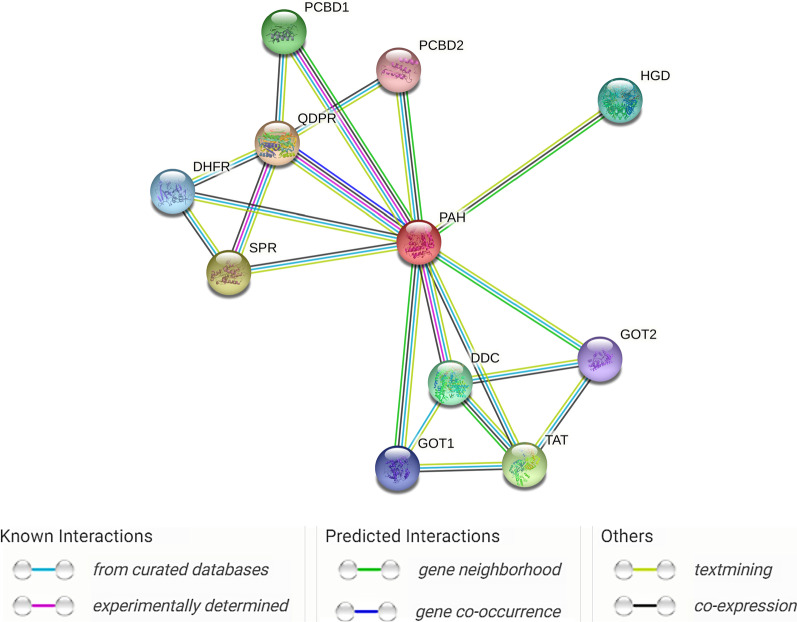
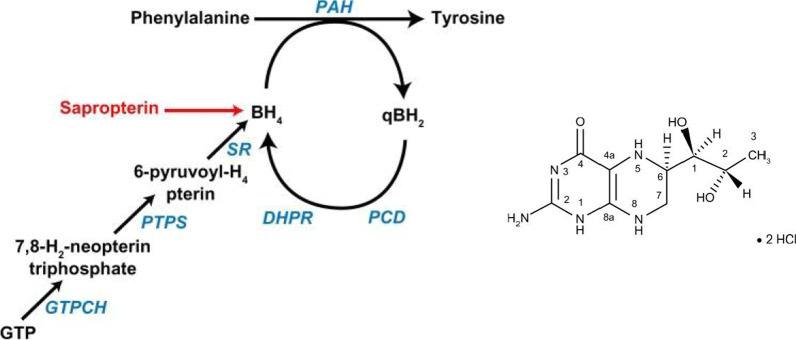
This review discusses the epidemiology, pathophysiology, genetic etiology, and management of phenylketonuria (PKU). PKU, an autosomal recessive disease, is an inborn error of phenylalanine (Phe) metabolism caused by pathogenic variants in the phenylalanine hydroxylase (PAH) gene. The prevalence of PKU varies widely among ethnicities and geographic regions, affecting approximately 1 in 24,000 individuals worldwide. Deficiency in the PAH enzyme or, in rare cases, the cofactor tetrahydrobiopterin results in high blood Phe concentrations, causing brain dysfunction. Untreated PKU, also known as PAH deficiency, results in severe and irreversible intellectual disability, epilepsy, behavioral disorders, and clinical features such as acquired microcephaly, seizures, psychological signs, and generalized hypopigmentation of skin (including hair and eyes). Severe phenotypes are classic PKU, and less severe forms of PAH deficiency are moderate PKU, mild PKU, mild hyperphenylalaninaemia (HPA), or benign HPA. Early diagnosis and intervention must start shortly after birth to prevent major cognitive and neurological effects. Dietary treatment, including natural protein restriction and Phe-free supplements, must be used to maintain blood Phe concentrations of 120-360 μmol/L throughout the life span. Additional treatments include the casein glycomacropeptide (GMP), which contains very limited aromatic amino acids and may improve immunological function, and large neutral amino acid (LNAA) supplementation to prevent plasma Phe transport into the brain. The synthetic BH4 analog, sapropterin hydrochloride (i.e., Kuvan®, BioMarin), is another potential treatment that activates residual PAH, thus decreasing Phe concentrations in the blood of PKU patients. Moreover, daily subcutaneous injection of pegylated Phe ammonia-lyase (i.e., pegvaliase; PALYNZIQ®, BioMarin) has promised gene therapy in recent clinical trials, and mRNA approaches are also being studied.
Keywords: Epidemiology; Genetic etiology; PKU management; Pathophysiology; Phenylalanine hydroxylase; Phenylketonuria; Tetrahydrobiopterin.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- Anikster Y, Haack TB, Vilboux T, Pode-Shakked B, Thony B, Shen N, Guarani V, Meissner T, Mayatepek E, Trefz FK, et al. Biallelic mutations in DNAJC12 cause hyperphenylalaninemia, dystonia, and intellectual disability. Am J Hum Genet. 2017;100(2):257–266. doi: 10.1016/j.ajhg.2017.01.002. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
