

SARS-CoV-2 infection in hamsters and humans results in lasting and unique systemic perturbations after recovery
- PMID: 35857629
- PMCID: PMC9210449
- DOI: 10.1126/scitranslmed.abq3059
SARS-CoV-2 infection in hamsters and humans results in lasting and unique systemic perturbations after recovery
Abstract
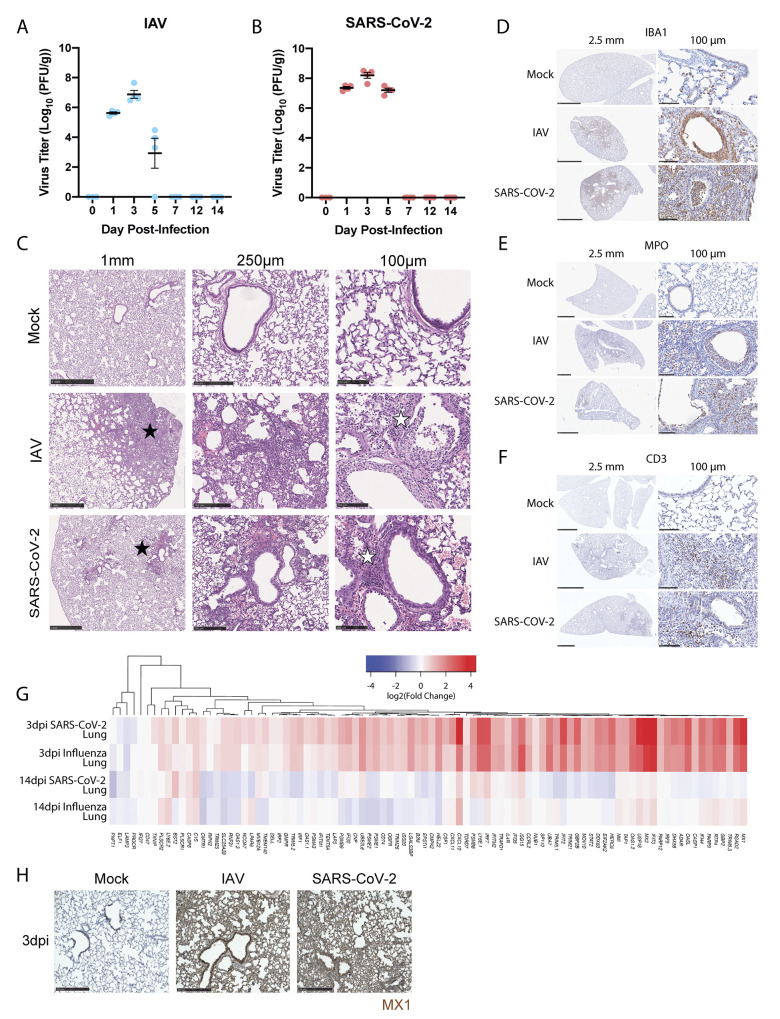
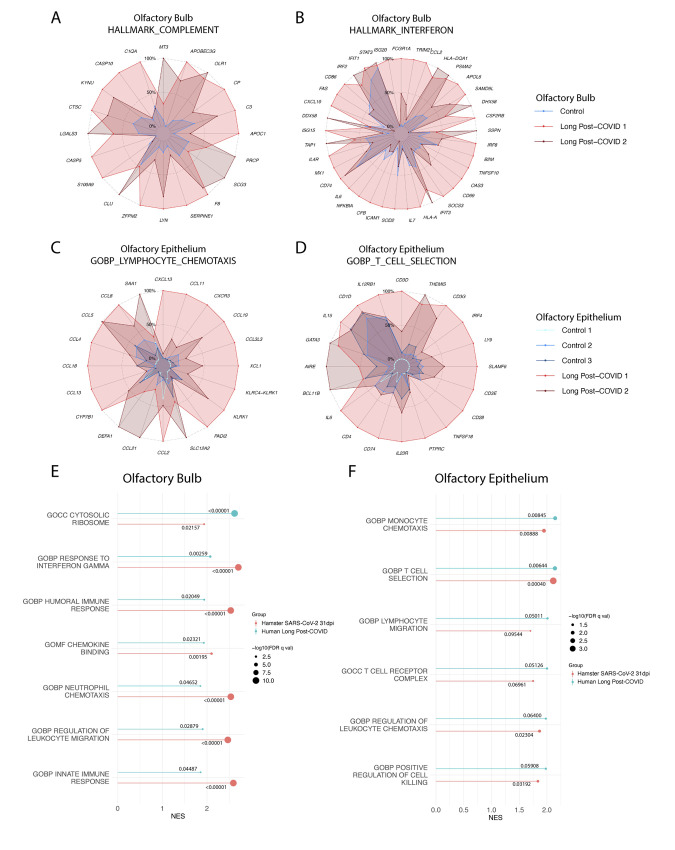
The host response to severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection can result in prolonged pathologies collectively referred to as post-acute sequalae of COVID-19 (PASC) or long COVID. To better understand the mechanism underlying long COVID biology, we compared the short- and long-term systemic responses in the golden hamster after either SARS-CoV-2 or influenza A virus (IAV) infection. Results demonstrated that SARS-CoV-2 exceeded IAV in its capacity to cause permanent injury to the lung and kidney and uniquely affected the olfactory bulb (OB) and olfactory epithelium (OE). Despite a lack of detectable infectious virus, the OB and OE demonstrated myeloid and T cell activation, proinflammatory cytokine production, and an interferon response that correlated with behavioral changes extending a month after viral clearance. These sustained transcriptional changes could also be corroborated from tissue isolated from individuals who recovered from COVID-19. These data highlight a molecular mechanism for persistent COVID-19 symptomology and provide a small animal model to explore future therapeutics.
Figures





References
-
- Zhu N., Zhang D., Wang W., Li X., Yang B., Song J., Zhao X., Huang B., Shi W., Lu R., Niu P., Zhan F., Ma X., Wang D., Xu W., Wu G., Gao G. F., Tan W.; China Novel Coronavirus Investigating and Research Team , A Novel Coronavirus from Patients with Pneumonia in China, 2019. N. Engl. J. Med. 382, 727–733 (2020). 10.1056/NEJMoa2001017 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
