

The protein organization of a red blood cell
- PMID: 35858567
- PMCID: PMC9764456
- DOI: 10.1016/j.celrep.2022.111103
The protein organization of a red blood cell
Abstract
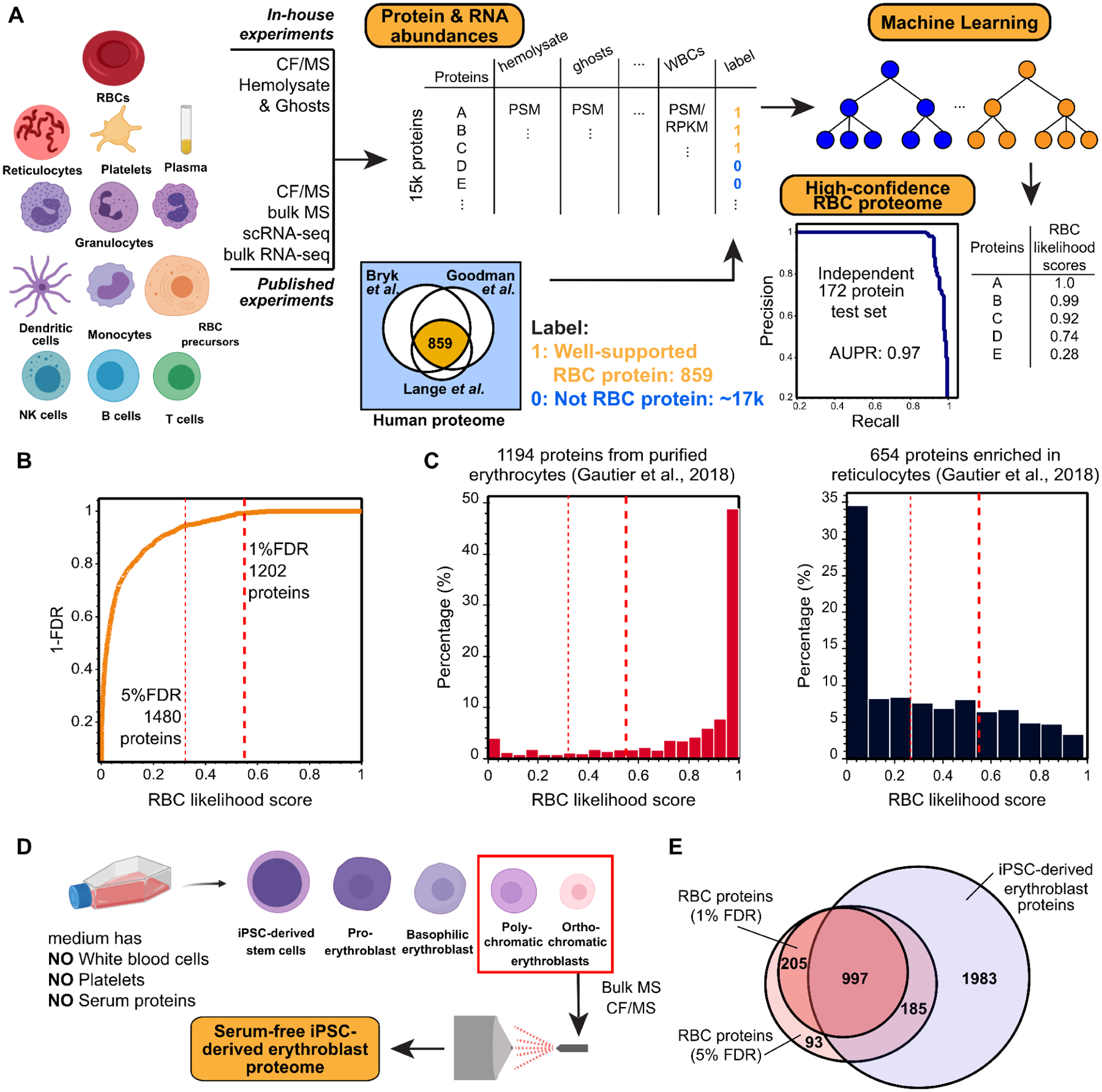
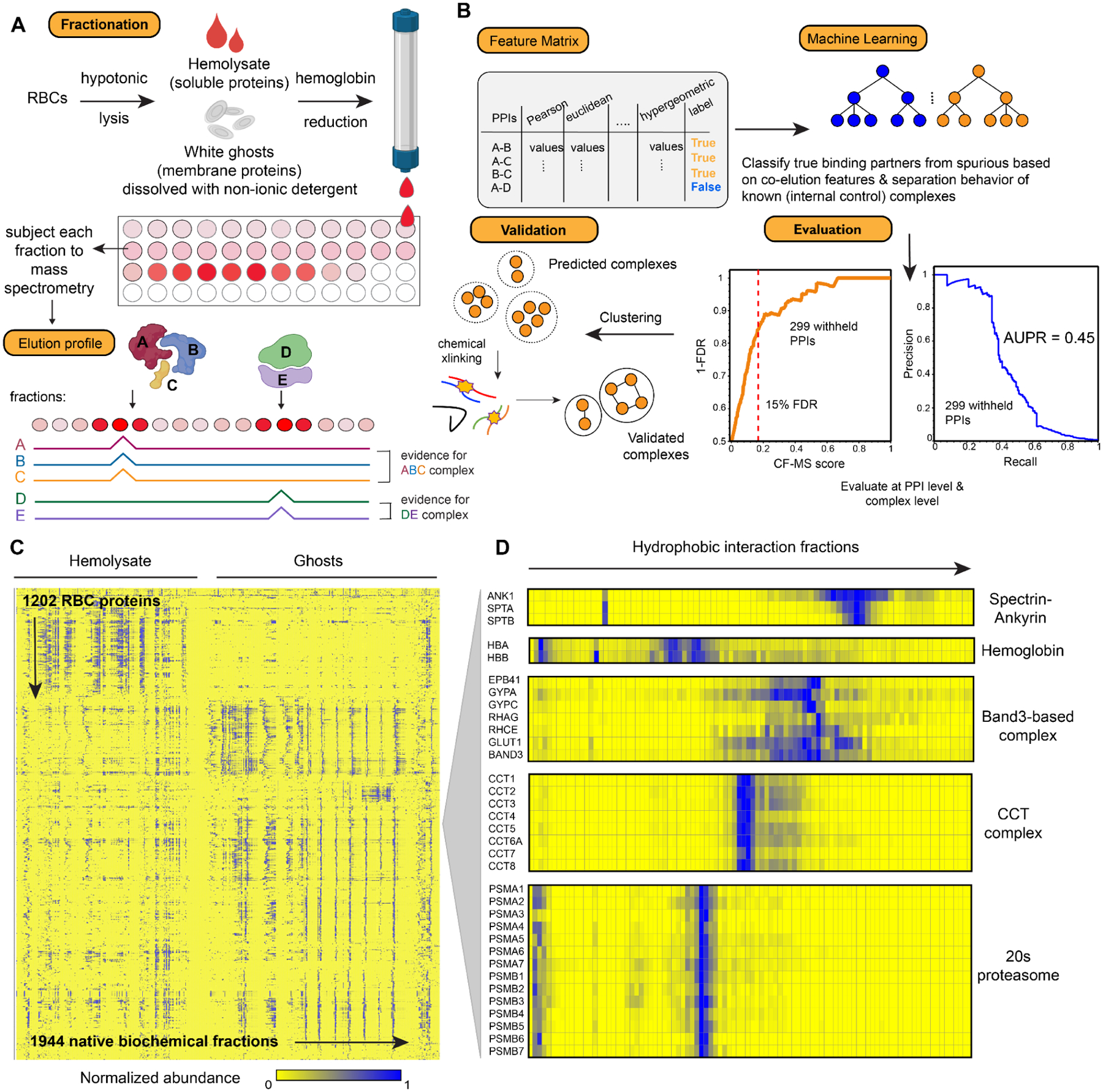
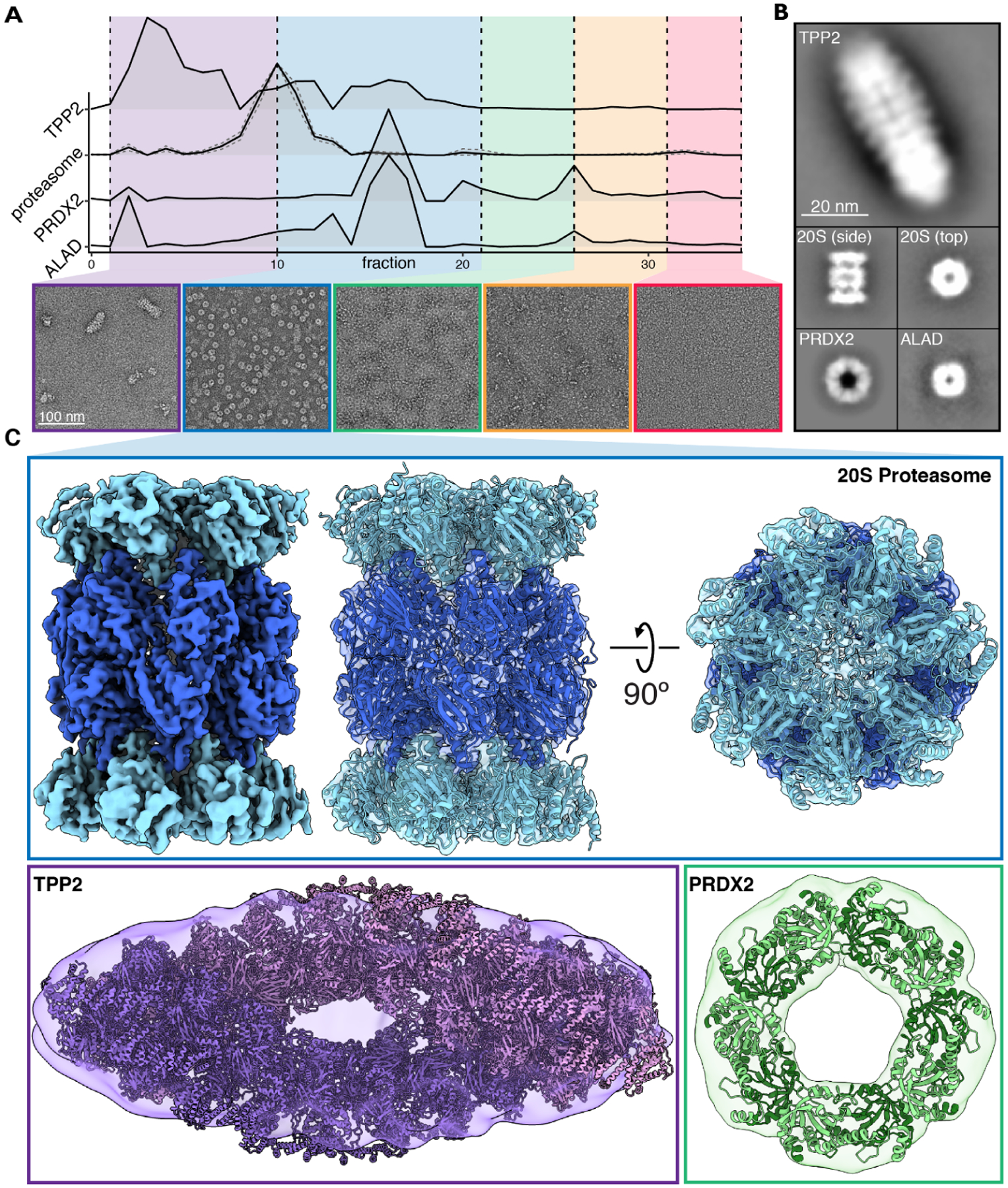
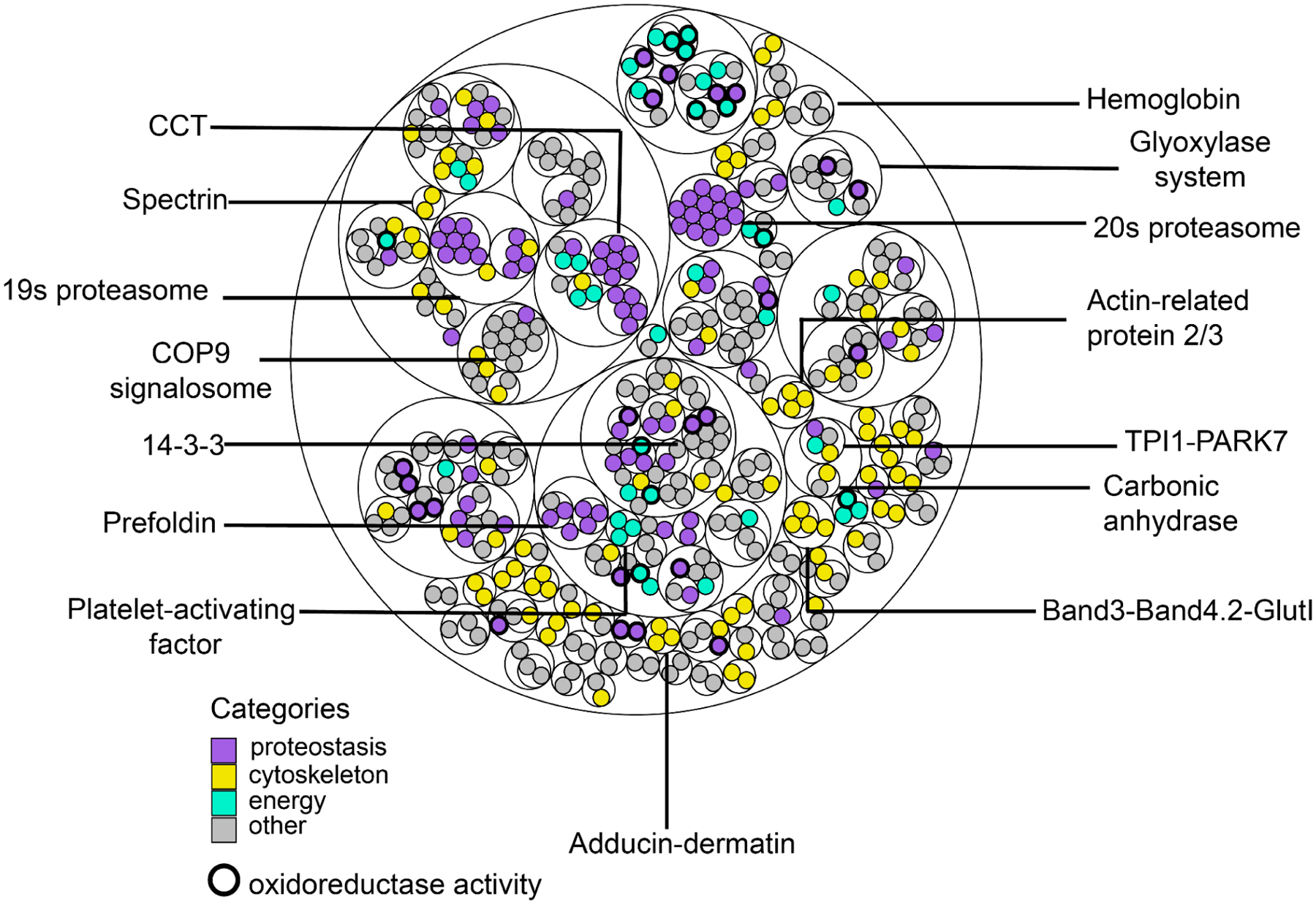
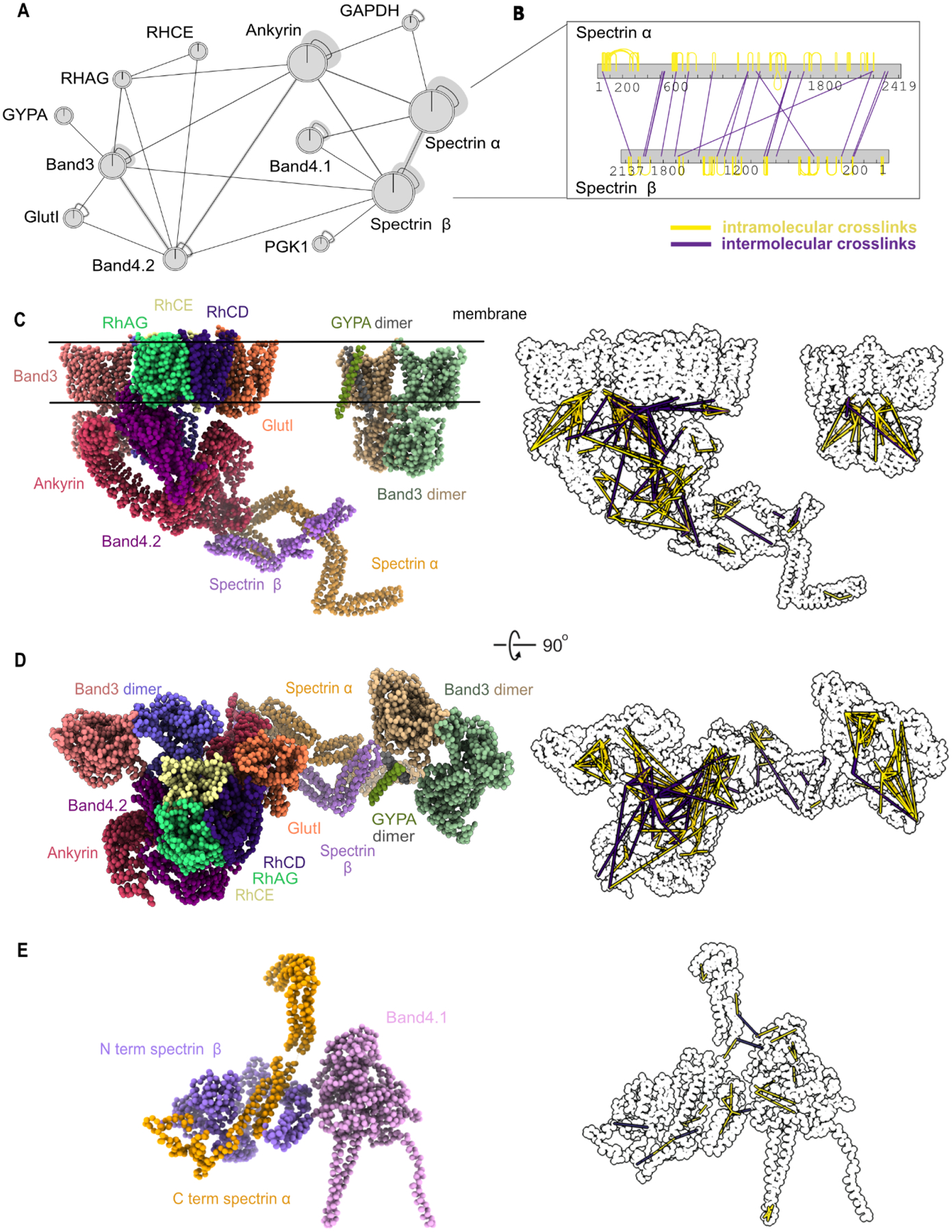
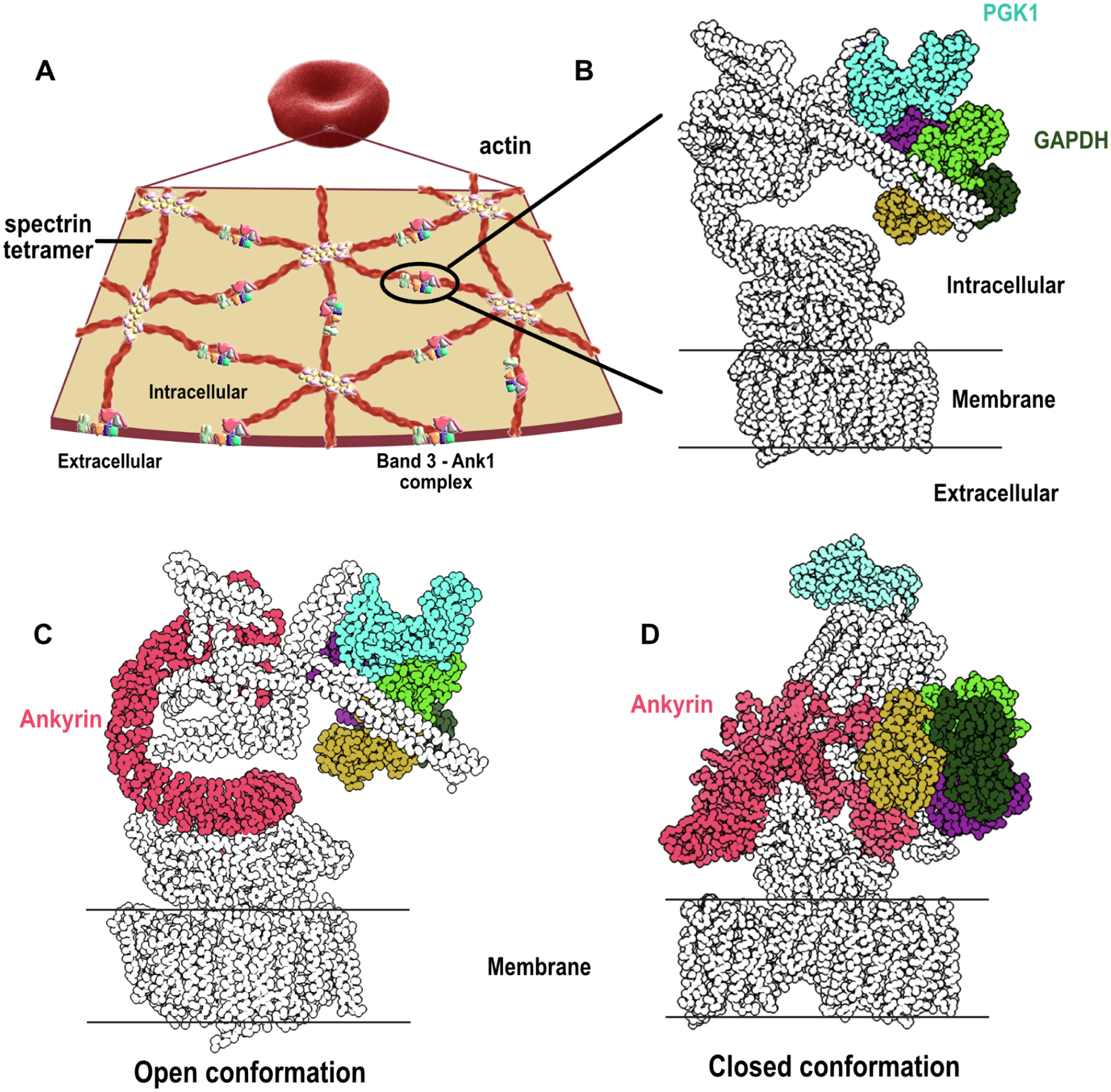
Red blood cells (RBCs) (erythrocytes) are the simplest primary human cells, lacking nuclei and major organelles and instead employing about a thousand proteins to dynamically control cellular function and morphology in response to physiological cues. In this study, we define a canonical RBC proteome and interactome using quantitative mass spectrometry and machine learning. Our data reveal an RBC interactome dominated by protein homeostasis, redox biology, cytoskeletal dynamics, and carbon metabolism. We validate protein complexes through electron microscopy and chemical crosslinking and, with these data, build 3D structural models of the ankyrin/Band 3/Band 4.2 complex that bridges the spectrin cytoskeleton to the RBC membrane. The model suggests spring-like compression of ankyrin may contribute to the characteristic RBC cell shape and flexibility. Taken together, our study provides an in-depth view of the global protein organization of human RBCs and serves as a comprehensive resource for future research.
Keywords: Band 3 complex; CP: Cell biology; erythrocytes; protein complexes; proteomics; red blood cells.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures
References
-
- Adams PD, Grosse-Kunstleve RW, Hung L-W, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr 58, 1948–1954. 10.1107/S0907444902016657. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
