

Pulse labeling reveals the tail end of protein folding by proteome profiling
- PMID: 35858568
- PMCID: PMC9893312
- DOI: 10.1016/j.celrep.2022.111096
Pulse labeling reveals the tail end of protein folding by proteome profiling
Abstract
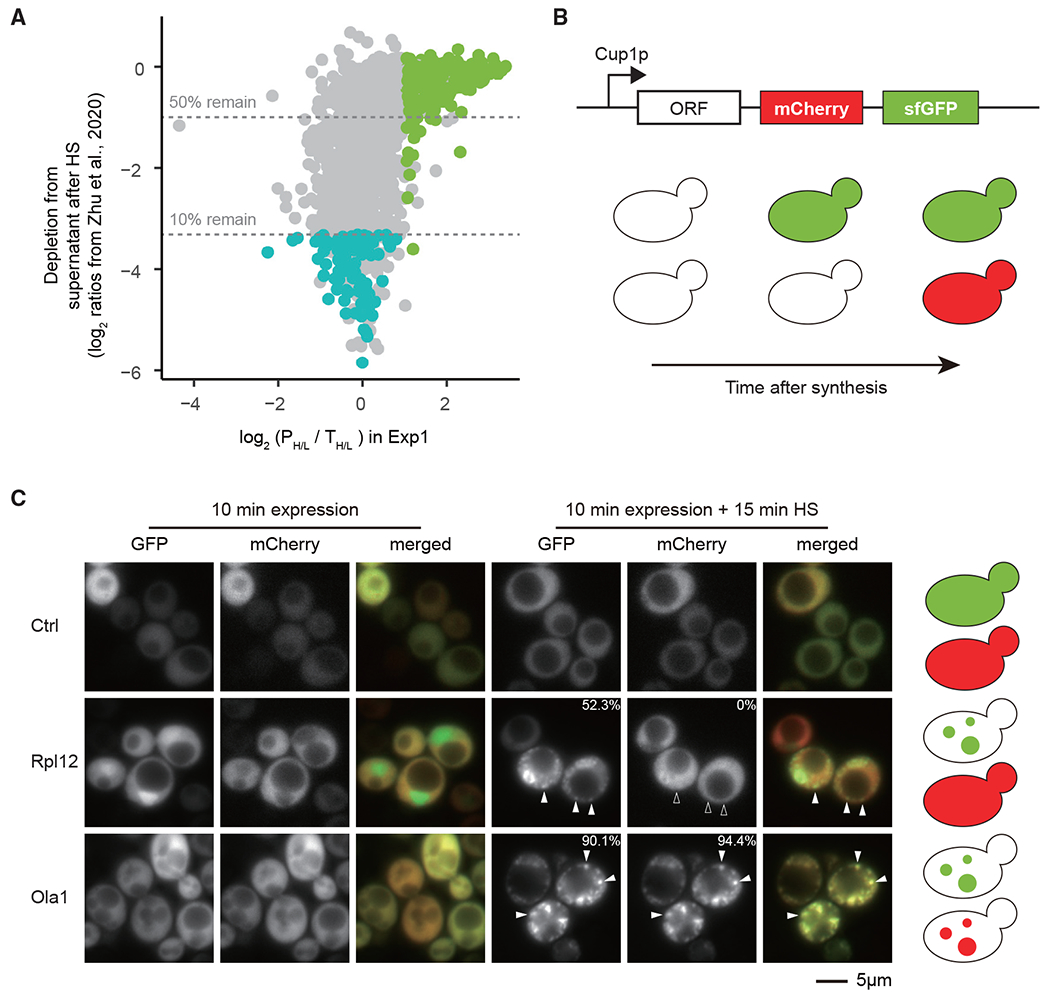
Accurate and efficient folding of nascent protein sequences into their native states requires support from the protein homeostasis network. Herein we probe which newly translated proteins are thermo-sensitive, making them susceptible to misfolding and aggregation under heat stress using pulse-SILAC mass spectrometry. We find a distinct group of proteins that is highly sensitive to this perturbation when newly synthesized but not once matured. These proteins are abundant and highly structured. Notably, they display a tendency to form β sheet secondary structures, have more complex folding topology, and are enriched for chaperone-binding motifs, suggesting a higher demand for chaperone-assisted folding. These polypeptides are also more often components of stable protein complexes in comparison with other proteins. Combining these findings suggests the existence of a specific subset of proteins in the cell that is particularly vulnerable to misfolding and aggregation following synthesis before reaching the native state.
Keywords: CP: Molecular biology; heat stress; limited proteolysis; misfolding; protein aggregation; protein folding; protein mass spectrometry; proteomics; proteostasis; pulse SILAC; translation.
Copyright © 2022 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures






Similar articles
-
Molecular chaperone functions in protein folding and proteostasis.Annu Rev Biochem. 2013;82:323-55. doi: 10.1146/annurev-biochem-060208-092442. Annu Rev Biochem. 2013. PMID: 23746257 Review.
-
Fluorescence Turn-On Folding Sensor To Monitor Proteome Stress in Live Cells.J Am Chem Soc. 2015 Sep 9;137(35):11303-11. doi: 10.1021/jacs.5b04366. Epub 2015 Aug 25. J Am Chem Soc. 2015. PMID: 26305239 Free PMC article.
-
Molecular chaperones in protein folding and proteostasis.Nature. 2011 Jul 20;475(7356):324-32. doi: 10.1038/nature10317. Nature. 2011. PMID: 21776078 Review.
-
Hsp110 cooperates with different cytosolic HSP70 systems in a pathway for de novo folding.J Biol Chem. 2005 Dec 16;280(50):41252-61. doi: 10.1074/jbc.M503615200. Epub 2005 Oct 11. J Biol Chem. 2005. PMID: 16219770
-
Conformational flexibility within the nascent polypeptide-associated complex enables its interactions with structurally diverse client proteins.J Biol Chem. 2018 Jun 1;293(22):8554-8568. doi: 10.1074/jbc.RA117.001568. Epub 2018 Apr 12. J Biol Chem. 2018. PMID: 29650757 Free PMC article.
Cited by
-
Shift of the insoluble content of the proteome in the aging mouse brain.Proc Natl Acad Sci U S A. 2023 Nov 7;120(45):e2310057120. doi: 10.1073/pnas.2310057120. Epub 2023 Oct 31. Proc Natl Acad Sci U S A. 2023. PMID: 37906643 Free PMC article.
-
How soluble misfolded proteins bypass chaperones at the molecular level.Nat Commun. 2023 Jun 21;14(1):3689. doi: 10.1038/s41467-023-38962-z. Nat Commun. 2023. PMID: 37344452 Free PMC article.
-
Entangled Motifs in Membrane Protein Structures.Int J Mol Sci. 2023 May 24;24(11):9193. doi: 10.3390/ijms24119193. Int J Mol Sci. 2023. PMID: 37298146 Free PMC article.
-
Non-covalent Lasso Entanglements in Folded Proteins: Prevalence, Functional Implications, and Evolutionary Significance.J Mol Biol. 2024 Mar 15;436(6):168459. doi: 10.1016/j.jmb.2024.168459. Epub 2024 Jan 30. J Mol Biol. 2024. PMID: 38296158 Free PMC article.
-
Folding kinetics of an entangled protein.PLoS Comput Biol. 2023 Nov 13;19(11):e1011107. doi: 10.1371/journal.pcbi.1011107. eCollection 2023 Nov. PLoS Comput Biol. 2023. PMID: 37956216 Free PMC article.
References
-
- Bauer DF, (1972). Constructing confidence sets using rank statistics. J. Am. Stat. Assoc 67, 687–690. 10.1080/01621459.1972.10481279. - DOI
-
- Beck S, Michalski A, Raether O, Lubeck M, Kaspar S, Goedecke N, Baessmann C, Hornburg D, Meier F, Paron I, et al. (2015). The impact II, a very high-resolution quadrupole time-of-flight instrument (QTOF) for deep shotgun proteomics. Mol. Cell. Proteomics 14, 2014–2029. 10.1074/mcp.m114.047407. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
