

Single-Cell Analysis Reveals the Range of Transcriptional States of Circulating Human Neutrophils
- PMID: 35858733
- PMCID: PMC9712146
- DOI: 10.4049/jimmunol.2200154
Single-Cell Analysis Reveals the Range of Transcriptional States of Circulating Human Neutrophils
Abstract
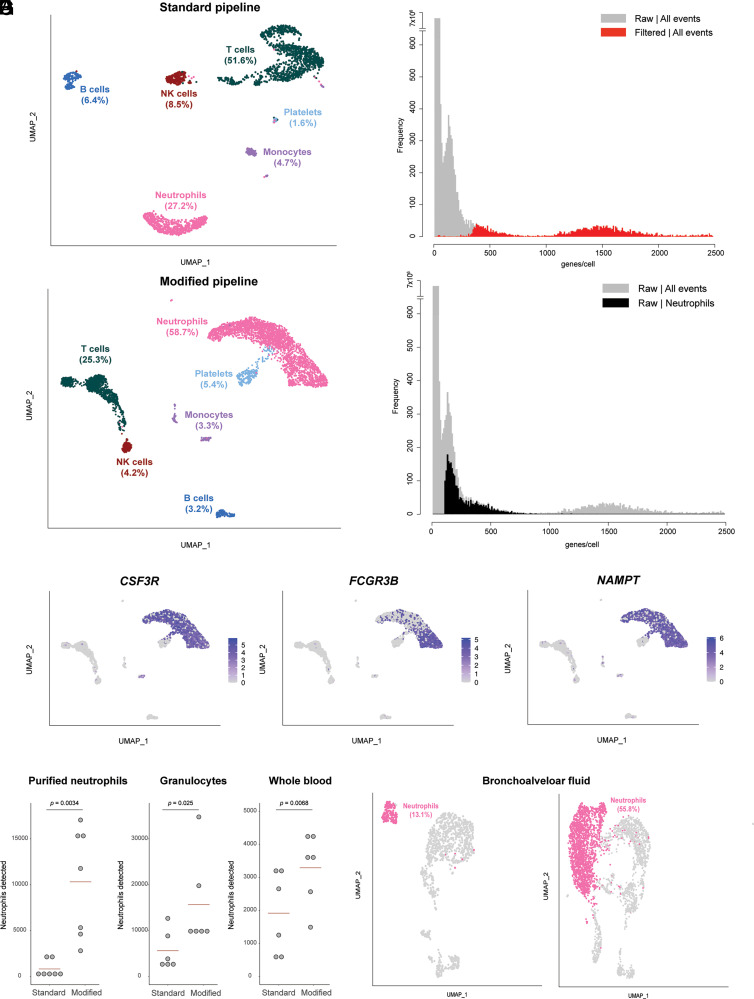
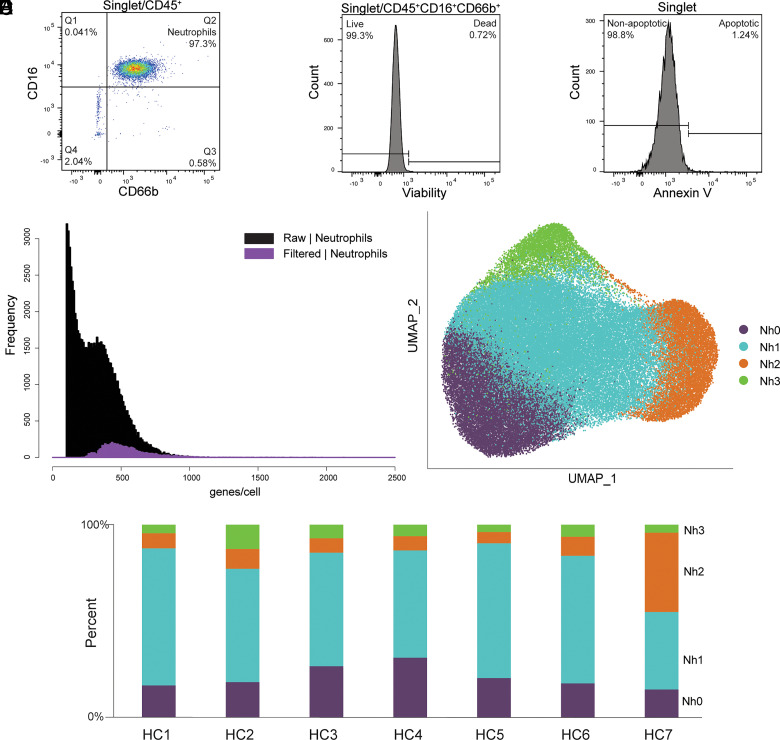
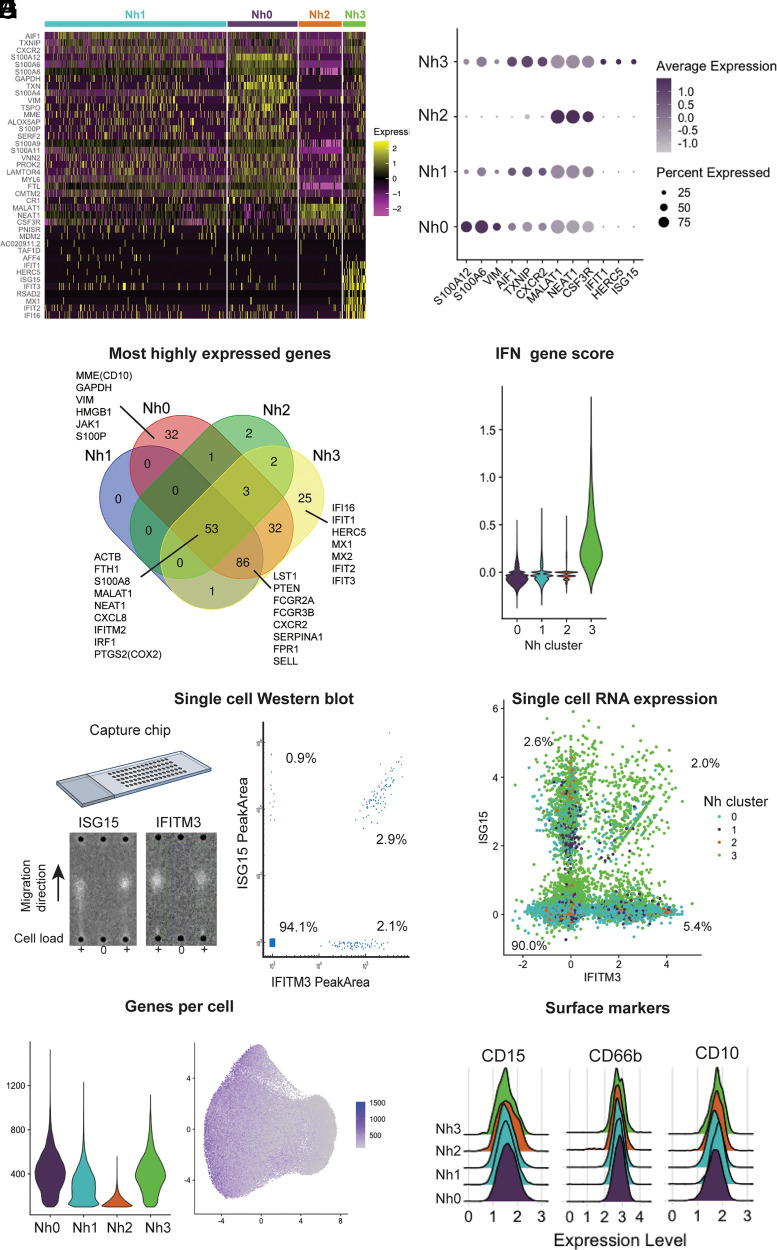
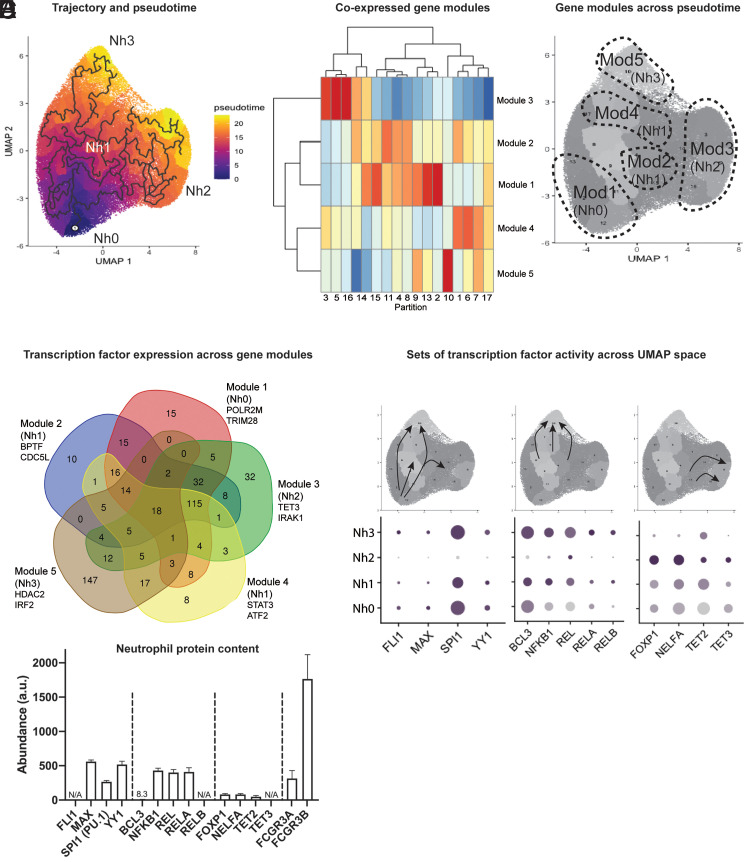
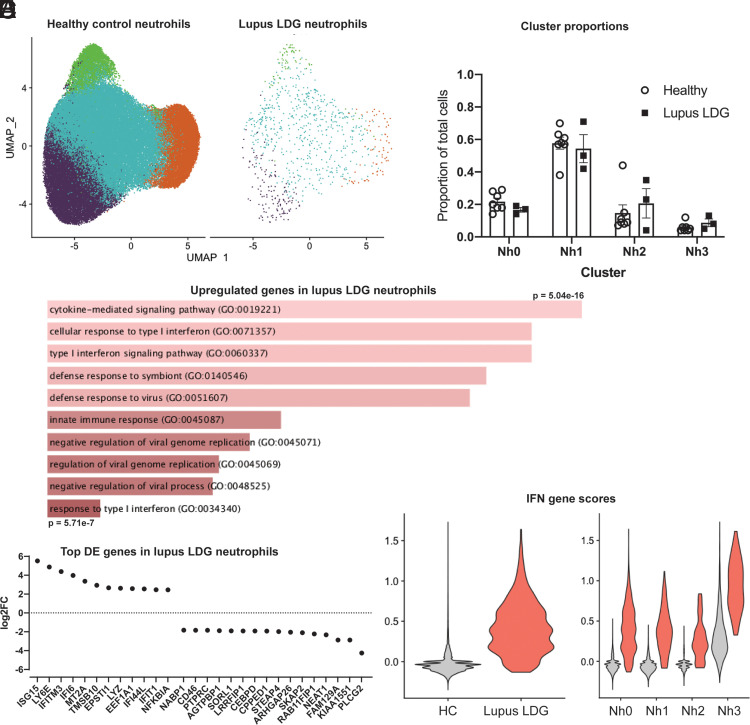
Neutrophils are the most abundant leukocytes in human blood and are essential components of innate immunity. Until recently, neutrophils were considered homogeneous and transcriptionally inactive cells, but both concepts are being challenged. Single-cell RNA sequencing (scRNA-seq) offers an unbiased view of cells along a continuum of transcriptional states. However, the use of scRNA-seq to characterize neutrophils has proven technically difficult, explaining in part the paucity of published single-cell data on neutrophils. We have found that modifications to the data analysis pipeline, rather than to the existing scRNA-seq chemistries, can significantly increase the detection of human neutrophils in scRNA-seq. We have then applied a modified pipeline to the study of human peripheral blood neutrophils. Our findings indicate that circulating human neutrophils are transcriptionally heterogeneous cells, which can be classified into one of four transcriptional clusters that are reproducible among healthy human subjects. We demonstrate that peripheral blood neutrophils shift from relatively immature (Nh0) cells, through a transitional phenotype (Nh1), into one of two end points defined by either relative transcriptional inactivity (Nh2) or high expression of type I IFN-inducible genes (Nh3). Transitions among states are characterized by the expression of specific transcription factors. By simultaneously measuring surface proteins and intracellular transcripts at the single-cell level, we show that these transcriptional subsets are independent of the canonical surface proteins that are commonly used to define and characterize human neutrophils. These findings provide a new view of human neutrophil heterogeneity, with potential implications for the characterization of neutrophils in health and disease.
Copyright © 2022 by The American Association of Immunologists, Inc.
Conflict of interest statement
The authors have no financial conflicts of interest.
Figures
References
-
- Ng L. G., Ostuni R., Hidalgo A.. 2019. Heterogeneity of neutrophils. Nat. Rev. Immunol. 19: 255–265. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
