

ATAC-STARR-seq reveals transcription factor-bound activators and silencers within chromatin-accessible regions of the human genome
- PMID: 35858748
- PMCID: PMC9435738
- DOI: 10.1101/gr.276766.122
ATAC-STARR-seq reveals transcription factor-bound activators and silencers within chromatin-accessible regions of the human genome
Abstract
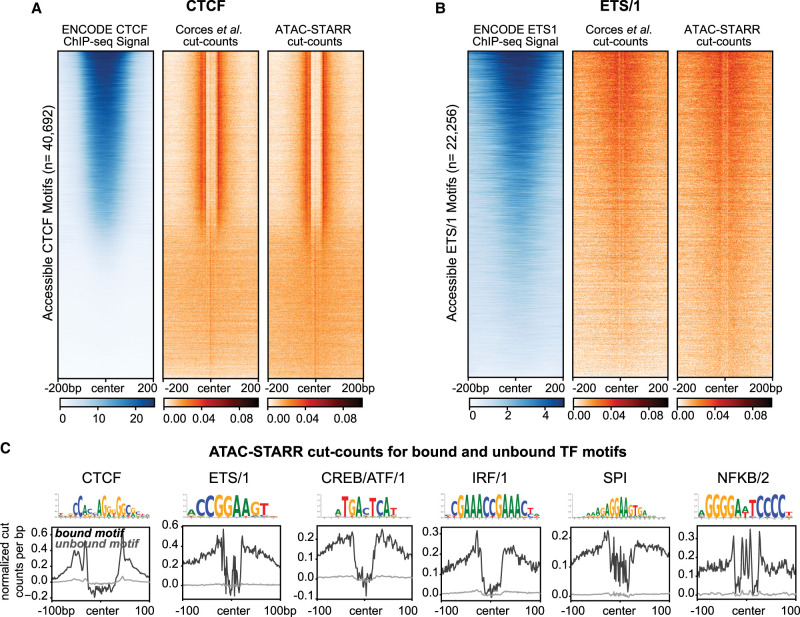
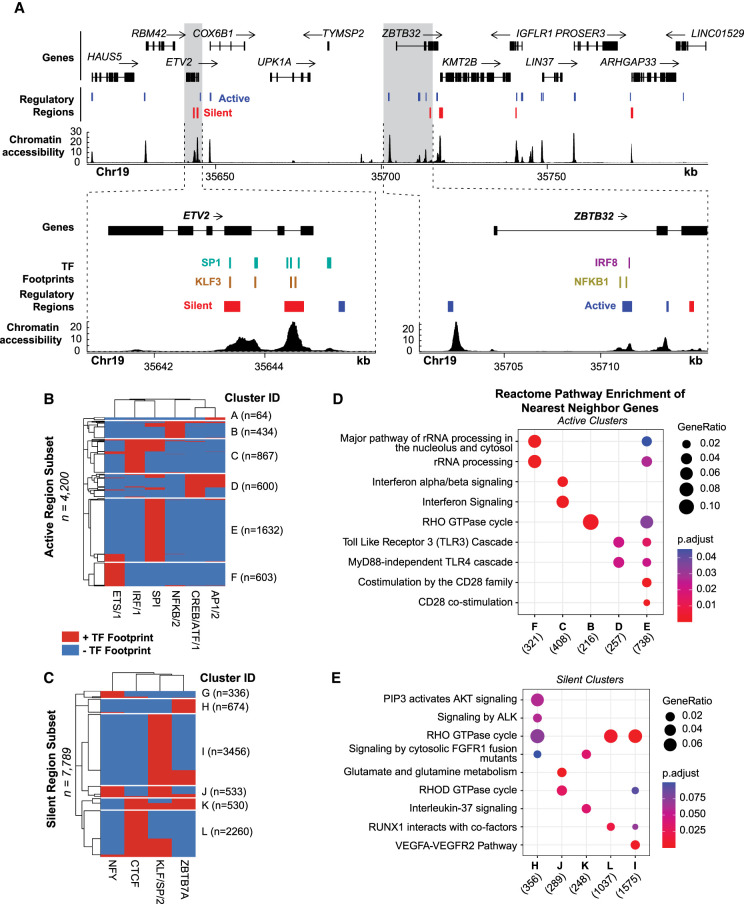
Massively parallel reporter assays (MPRAs) test the capacity of putative gene regulatory elements to drive transcription on a genome-wide scale. Most gene regulatory activity occurs within accessible chromatin, and recently described methods have combined assays that capture these regions-such as assay for transposase-accessible chromatin using sequencing (ATAC-seq)-with self-transcribing active regulatory region sequencing (STARR-seq) to selectively assay the regulatory potential of accessible DNA (ATAC-STARR-seq). Here, we report an integrated approach that quantifies activating and silencing regulatory activity, chromatin accessibility, and transcription factor (TF) occupancy with one assay using ATAC-STARR-seq. Our strategy, including important updates to the ATAC-STARR-seq assay and workflow, enabled high-resolution testing of ∼50 million unique DNA fragments tiling ∼101,000 accessible chromatin regions in human lymphoblastoid cells. We discovered that 30% of all accessible regions contain an activator, a silencer, or both. Although few MPRA studies have explored silencing activity, we demonstrate that silencers occur at similar frequencies to activators, and they represent a distinct functional group enriched for unique TF motifs and repressive histone modifications. We further show that Tn5 cut-site frequencies are retained in the ATAC-STARR plasmid library compared to standard ATAC-seq, enabling TF occupancy to be ascertained from ATAC-STARR data. With this approach, we found that activators and silencers cluster by distinct TF footprint combinations, and these groups of activity represent different gene regulatory networks of immune cell function. Altogether, these data highlight the multilayered capabilities of ATAC-STARR-seq to comprehensively investigate the regulatory landscape of the human genome all from a single DNA fragment source.
© 2022 Hansen and Hodges; Published by Cold Spring Harbor Laboratory Press.
Figures
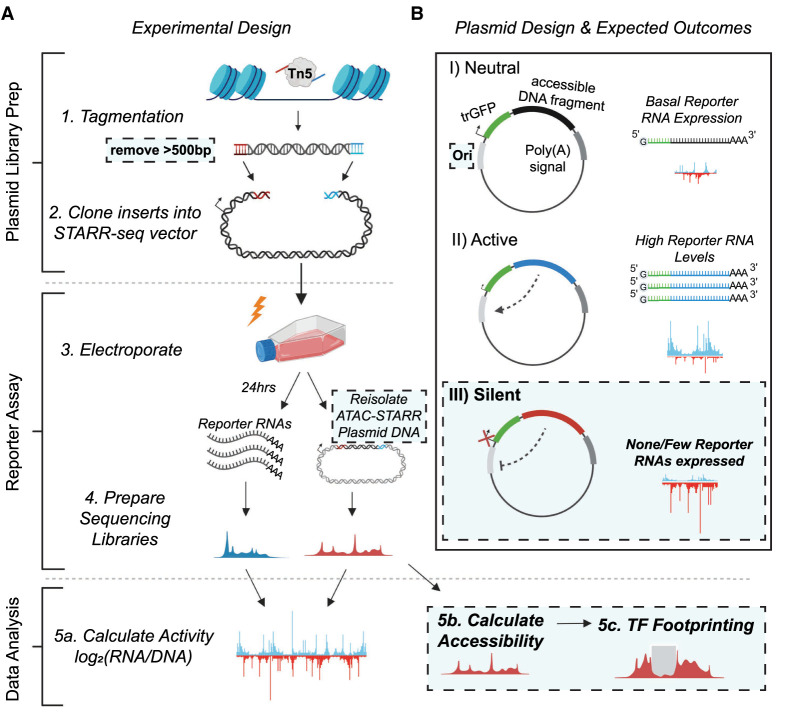
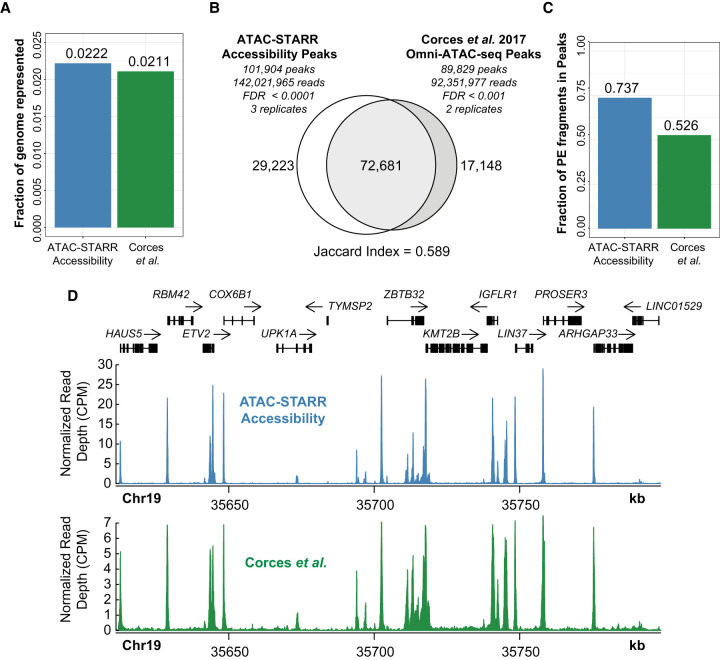
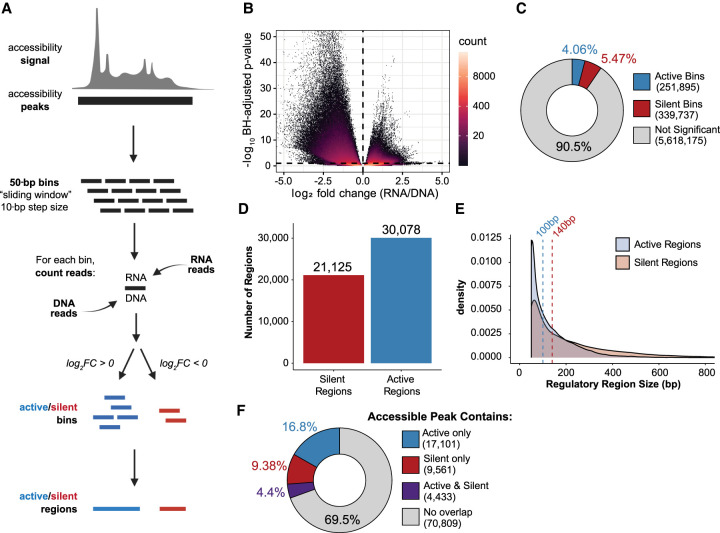
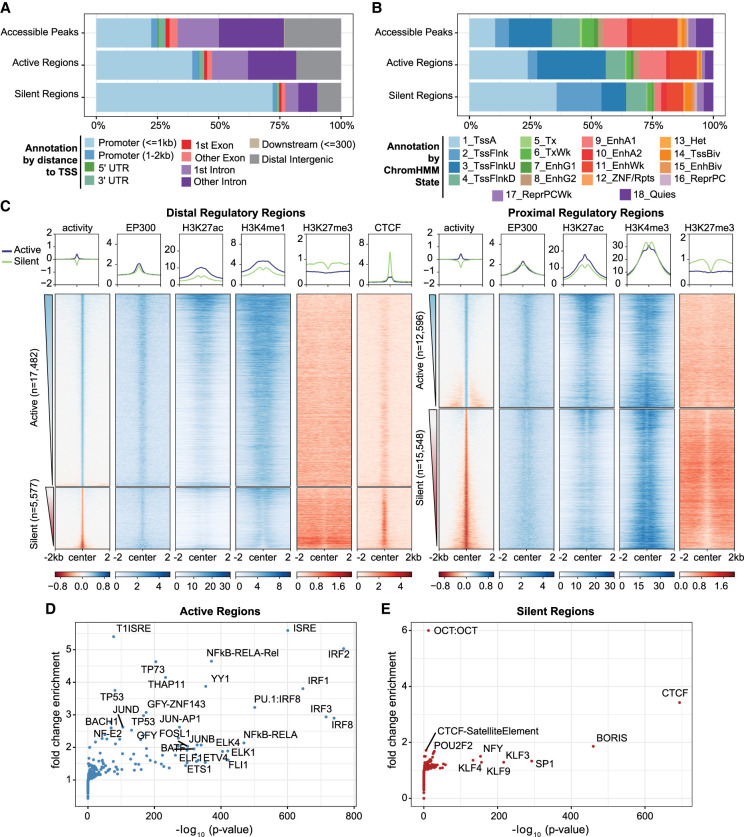
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous