

Polycomb-lamina antagonism partitions heterochromatin at the nuclear periphery
- PMID: 35859152
- PMCID: PMC9300685
- DOI: 10.1038/s41467-022-31857-5
Polycomb-lamina antagonism partitions heterochromatin at the nuclear periphery
Abstract
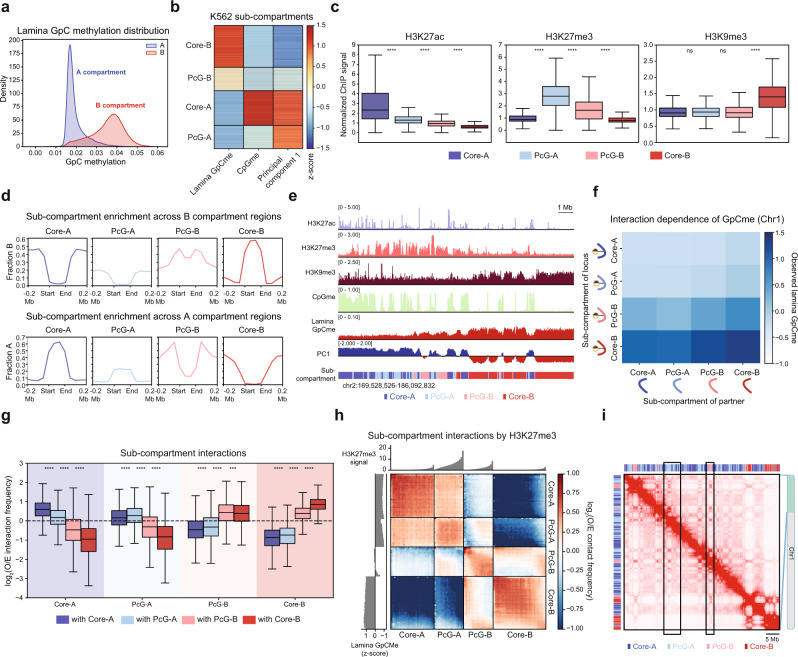
The genome can be divided into two spatially segregated compartments, A and B, which partition active and inactive chromatin states. While constitutive heterochromatin is predominantly located within the B compartment near the nuclear lamina, facultative heterochromatin marked by H3K27me3 spans both compartments. How epigenetic modifications, compartmentalization, and lamina association collectively maintain heterochromatin architecture remains unclear. Here we develop Lamina-Inducible Methylation and Hi-C (LIMe-Hi-C) to jointly measure chromosome conformation, DNA methylation, and lamina positioning. Through LIMe-Hi-C, we identify topologically distinct sub-compartments with high levels of H3K27me3 and differing degrees of lamina association. Inhibition of Polycomb repressive complex 2 (PRC2) reveals that H3K27me3 is essential for sub-compartment segregation. Unexpectedly, PRC2 inhibition promotes lamina association and constitutive heterochromatin spreading into H3K27me3-marked B sub-compartment regions. Consistent with this repositioning, genes originally marked with H3K27me3 in the B compartment, but not the A compartment, remain largely repressed, suggesting that constitutive heterochromatin spreading can compensate for H3K27me3 loss at a transcriptional level. These findings demonstrate that Polycomb sub-compartments and their antagonism with lamina association are fundamental features of genome structure. More broadly, by jointly measuring nuclear position and Hi-C contacts, our study demonstrates how compartmentalization and lamina association represent distinct but interdependent modes of heterochromatin regulation.
© 2022. The Author(s).
Conflict of interest statement
B.B.L. is on the Scientific Advisory Board of H3 Biomedicine. M.J.A. declares outside interest in Excelsior Genomics. The remaining authors declare no competing interests.
Figures





References
-
- Kempfer, R. & Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet21, 207–226 (2019). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
