

Predicting and explaining the impact of genetic disruptions and interactions on organismal viability
- PMID: 35861390
- PMCID: PMC9438956
- DOI: 10.1093/bioinformatics/btac519
Predicting and explaining the impact of genetic disruptions and interactions on organismal viability
Abstract
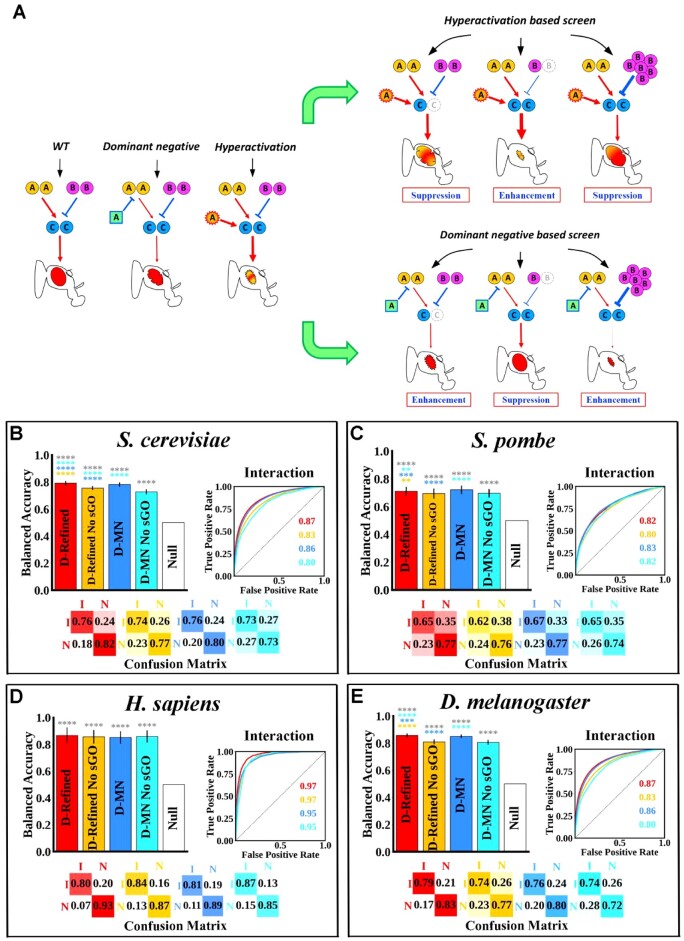
Motivation: Existing computational models can predict single- and double-mutant fitness but they do have limitations. First, they are often tested via evaluation metrics that are inappropriate for imbalanced datasets. Second, all of them only predict a binary outcome (viable or not, and negatively interacting or not). Third, most are uninterpretable black box machine learning models.
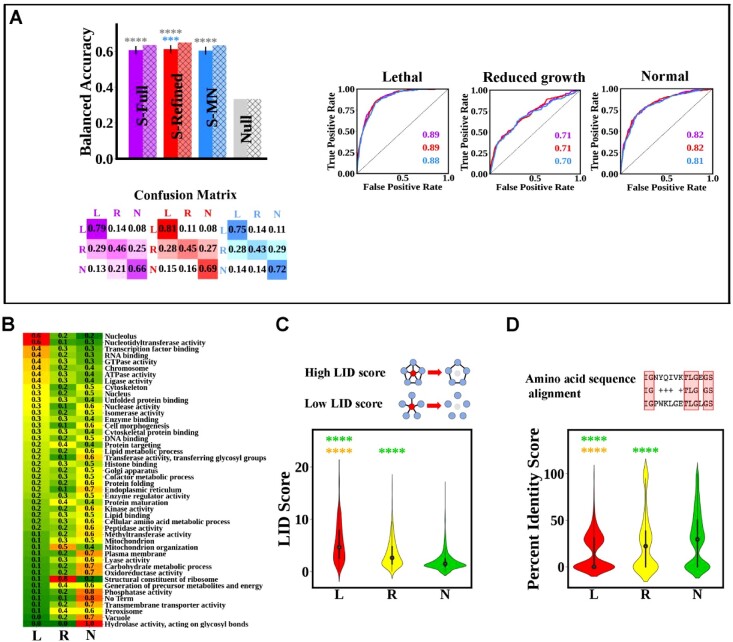
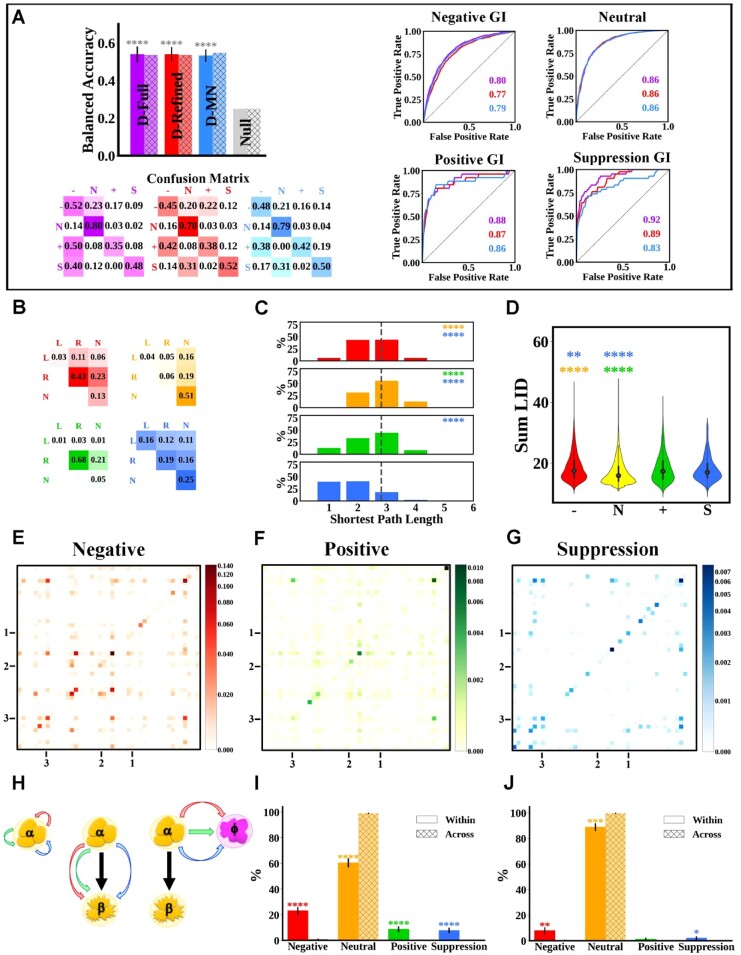
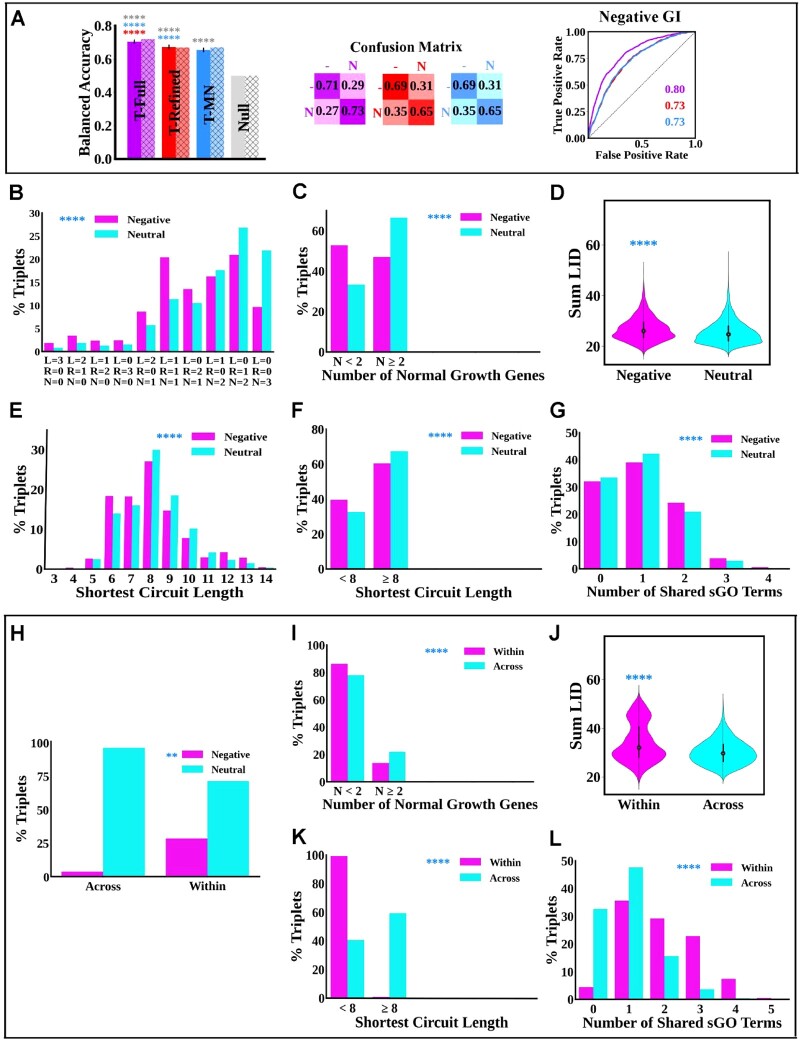
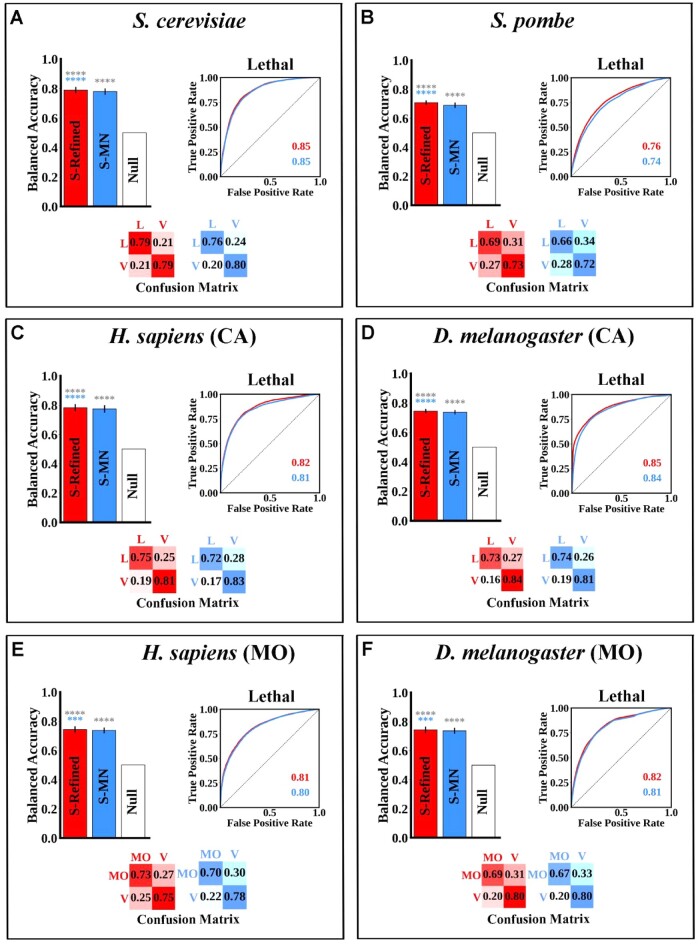
Results: Budding yeast datasets were used to develop high-performance Multinomial Regression (MN) models capable of predicting the impact of single, double and triple genetic disruptions on viability. These models are interpretable and give realistic non-binary predictions and can predict negative genetic interactions (GIs) in triple-gene knockouts. They are based on a limited set of gene features and their predictions are influenced by the probability of target gene participating in molecular complexes or pathways. Furthermore, the MN models have utility in other organisms such as fission yeast, fruit flies and humans, with the single gene fitness MN model being able to distinguish essential genes necessary for cell-autonomous viability from those required for multicellular survival. Finally, our models exceed the performance of previous models, without sacrificing interpretability.
Availability and implementation: All code and processed datasets used to generate results and figures in this manuscript are available at our Github repository at https://github.com/KISRDevelopment/cell_viability_paper. The repository also contains a link to the GI prediction website that lets users search for GIs using the MN models.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2022. Published by Oxford University Press.
Figures
Similar articles
-
Expression-based prediction of human essential genes and candidate lncRNAs in cancer cells.Bioinformatics. 2021 Apr 20;37(3):396-403. doi: 10.1093/bioinformatics/btaa717. Bioinformatics. 2021. PMID: 32790840
-
LinkExplorer: predicting, explaining and exploring links in large biomedical knowledge graphs.Bioinformatics. 2022 Apr 12;38(8):2371-2373. doi: 10.1093/bioinformatics/btac068. Bioinformatics. 2022. PMID: 35139158
-
Predictive and interpretable models via the stacked elastic net.Bioinformatics. 2021 Aug 4;37(14):2012-2016. doi: 10.1093/bioinformatics/btaa535. Bioinformatics. 2021. PMID: 32437519 Free PMC article.
-
Benchmarking of Machine Learning classifiers on plasma proteomic for COVID-19 severity prediction through interpretable artificial intelligence.Artif Intell Med. 2023 Mar;137:102490. doi: 10.1016/j.artmed.2023.102490. Epub 2023 Jan 18. Artif Intell Med. 2023. PMID: 36868685 Free PMC article. Review.
-
Insights into performance evaluation of compound-protein interaction prediction methods.Bioinformatics. 2022 Sep 16;38(Suppl_2):ii75-ii81. doi: 10.1093/bioinformatics/btac496. Bioinformatics. 2022. PMID: 36124806
Cited by
-
Complex synthetic lethality in cancer.Nat Genet. 2023 Dec;55(12):2039-2048. doi: 10.1038/s41588-023-01557-x. Epub 2023 Nov 30. Nat Genet. 2023. PMID: 38036785 Review.
References
-
- Alberts B. et al. (2002). Molecular Biology of the Cell. Garland Science, New York.
-
- Babu M.M. et al. (2004) Structure and evolution of transcriptional regulatory networks. Curr. Opin. Struct. Biol., 14, 283–291. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
