

Intercellular model predicts mechanisms of inflammation-fibrosis coupling after myocardial infarction
- PMID: 35862254
- PMCID: PMC9859968
- DOI: 10.1113/JP283346
Intercellular model predicts mechanisms of inflammation-fibrosis coupling after myocardial infarction
Abstract
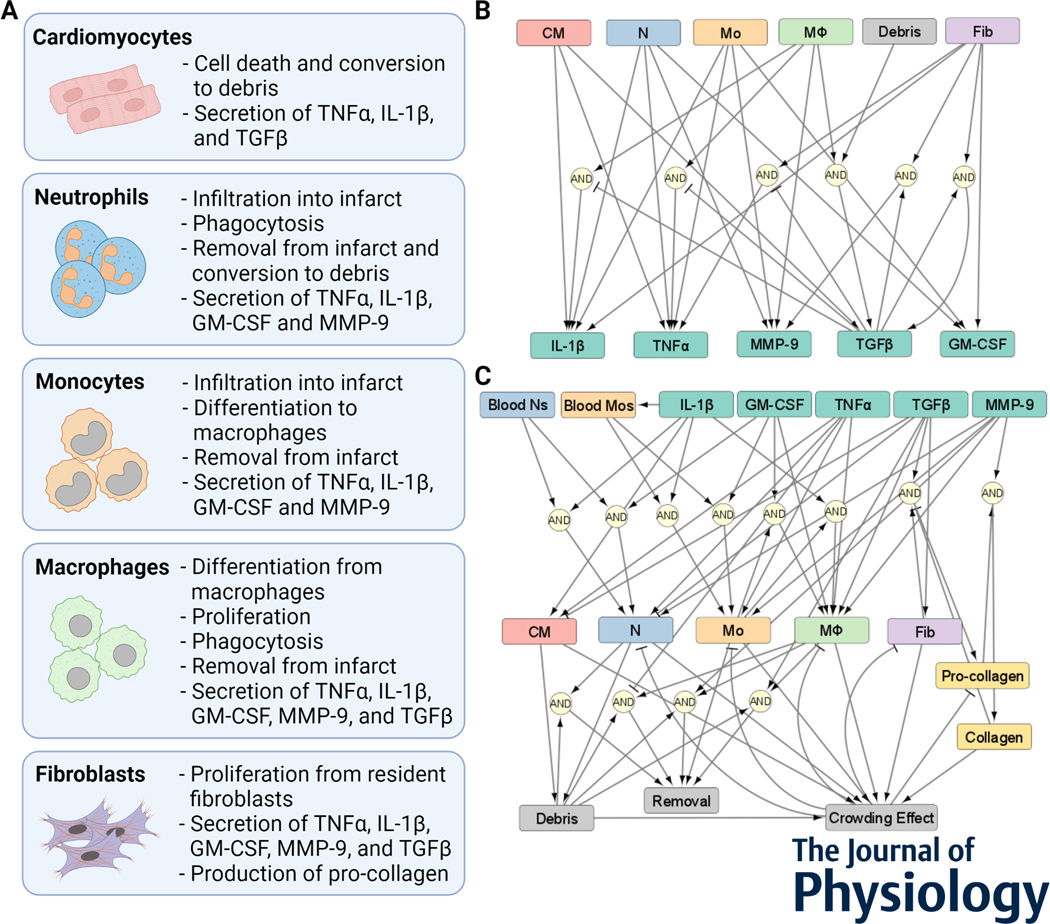
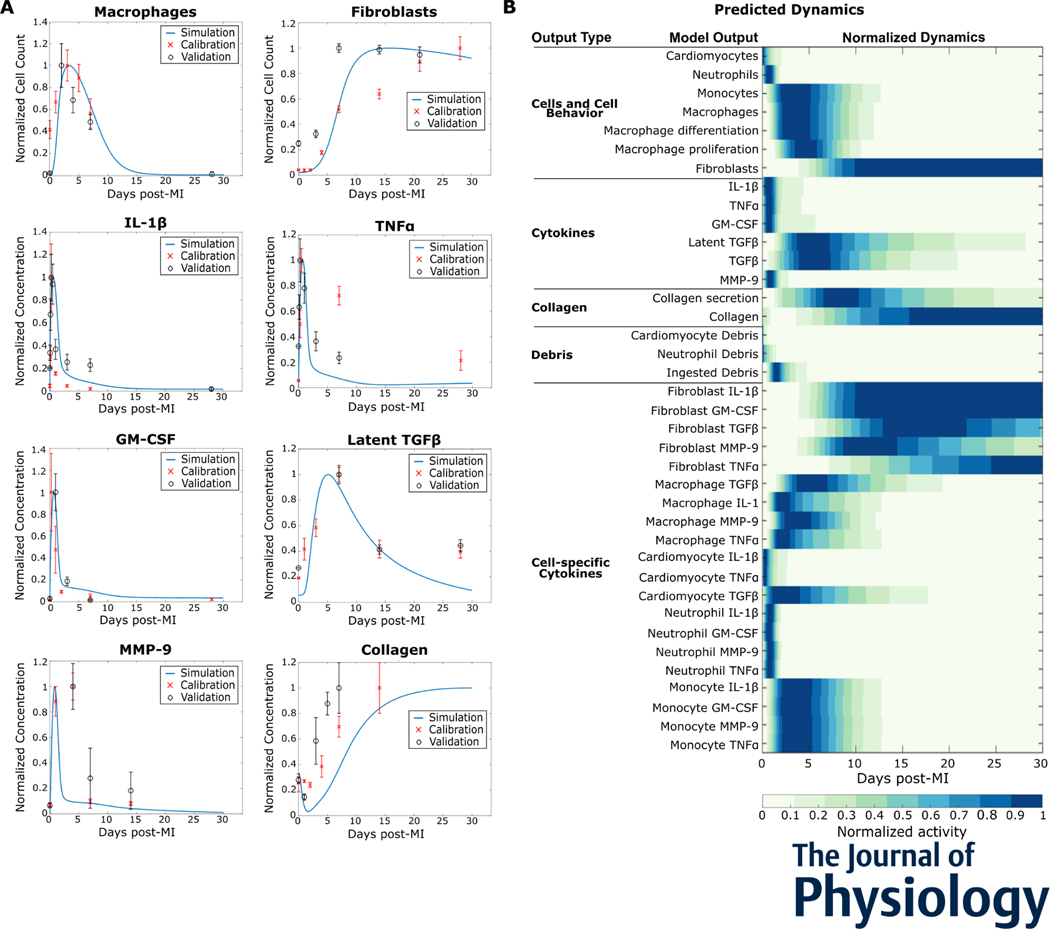
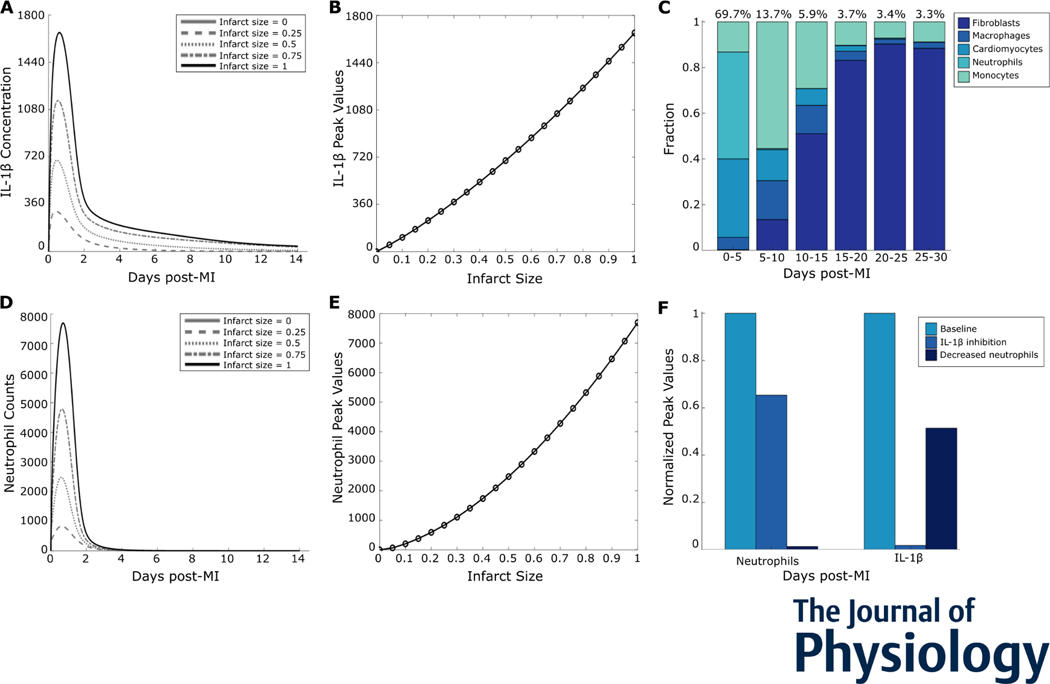
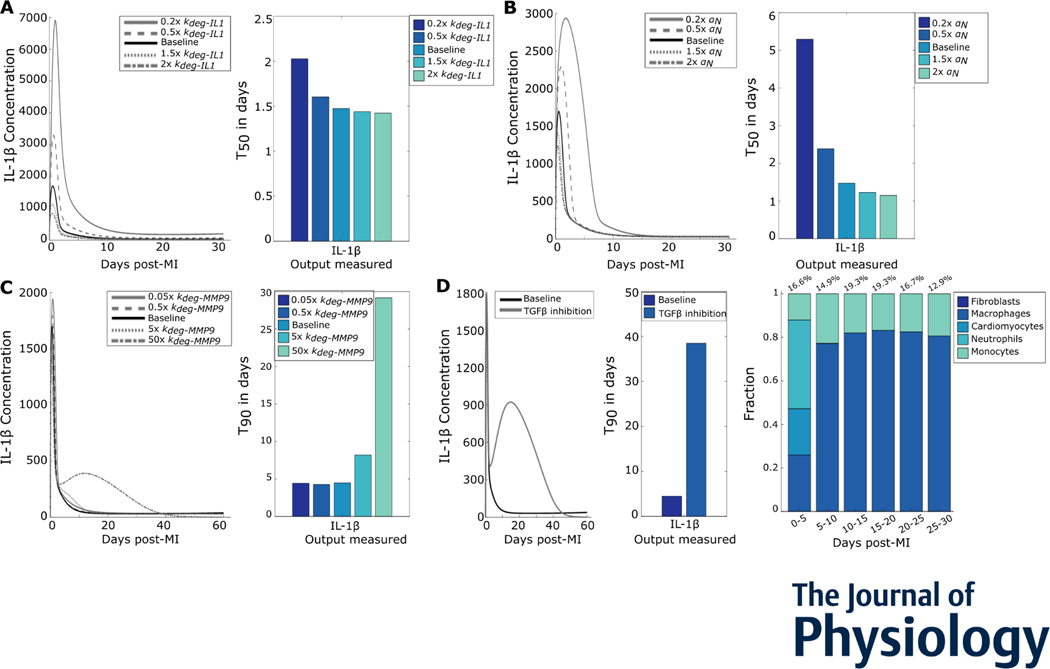
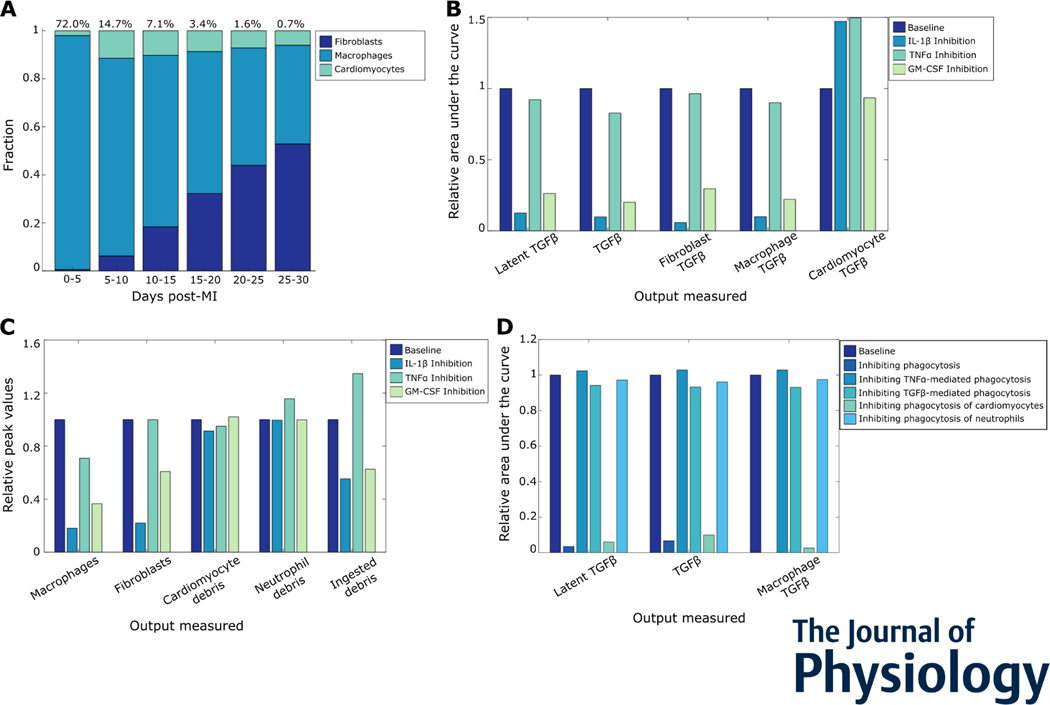
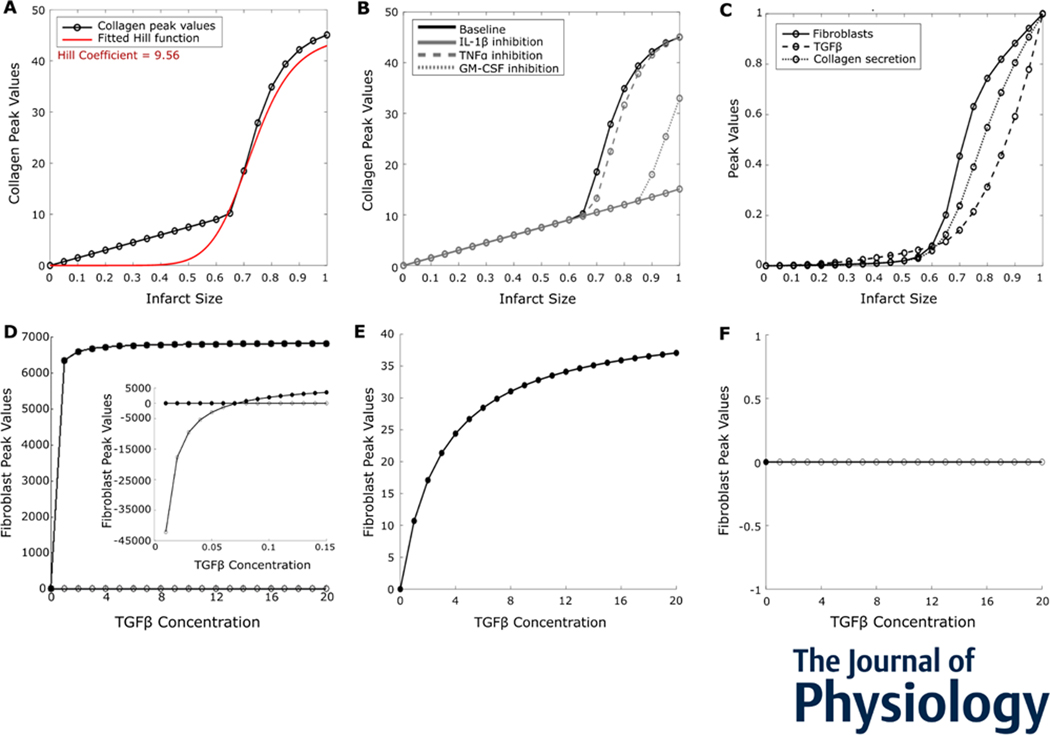
After myocardial infarction (MI), cardiac cells work together to regulate wound healing of the infarct. The pathological response to MI yields cardiac remodelling comprising inflammatory and fibrosis phases, and the interplay of cellular dynamics that underlies these phases has not been elucidated. This study developed a computational model to identify cytokine and cellular dynamics post-MI to predict mechanisms driving post-MI inflammation, resolution of inflammation, and scar formation. Additionally, this study evaluated the interdependence between inflammation and fibrosis. Our model bypassed limitations of in vivo approaches in achieving cellular specificity and performing specific perturbations such as global knockouts of chemical factors. The model predicted that inflammation is a graded response to initial infarct size that is amplified by a positive feedback loop between neutrophils and interleukin 1β (IL-1β). Resolution of inflammation was driven by degradation of IL-1β, matrix metalloproteinase 9, and transforming growth factor β (TGF-β), as well as apoptosis of neutrophils. Inflammation regulated TGFβ secretion directly through immune cell recruitment and indirectly through upregulation of macrophage phagocytosis. Lastly, we found that mature collagen deposition was an ultrasensitive switch in response to inflammation, which was amplified primarily by cardiac fibroblast proliferation. These findings describe the relationship between inflammation and fibrosis and highlight how the two responses work together post-MI. This model revealed that post-MI inflammation and fibrosis are dynamically coupled, which provides rationale for designing novel anti-inflammatory, pro-resolving or anti-fibrotic therapies that may improve the response to MI. KEY POINTS: Inflammation and matrix remodelling are two processes involved in wound healing after a heart attack. Cardiac cells work together to facilitate these processes; this is done by secreting cytokines that then regulate the cells themselves or other cells surrounding them. This study developed a computational model of the dynamics of cardiac cells and cytokines to predict mechanisms through which inflammation and matrix remodelling is regulated. We show the roles of various cytokines and signalling motifs in driving inflammation, resolution of inflammation and fibrosis. The novel concept of inflammation-fibrosis coupling, based on the model prediction that inflammation and fibrosis are dynamically coupled, provides rationale for future studies and for designing therapeutics to improve the response after a heart attack.
Keywords: inflammation-fibrosis coupling; intercellular dynamics; myocardial infarction.
© 2022 The Authors. The Journal of Physiology © 2022 The Physiological Society.
Figures

References
-
- Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GGL, Houser J-E, Qureshi IZ, Ownby ED, Gustini E, Biasucci LM, Severino A, Capogrossi MC, Vetrovec GW, Crea F, Baldi A, Kukreja RC, & Dobrina A. (2008). Anakinra, a Recombinant Human Interleukin-1 Receptor Antagonist, Inhibits Apoptosis in Experimental Acute Myocardial Infarction. Circulation, 117(20), 2670–2683. 10.1161/CIRCULATIONAHA.107.740233 - DOI - PubMed
-
- Al-Darraji A, Haydar D, Chelvarajan L, Tripathi H, Levitan B, Gao E, Venditto VJ, Gensel JC, Feola DJ, & Abdel-Latif A. (2018). Azithromycin therapy reduces cardiac inflammation and mitigates adverse cardiac remodeling after myocardial infarction: Potential therapeutic targets in ischemic heart disease. PloS One, 13(7), e0200474. 10.1371/journal.pone.0200474 - DOI - PMC - PubMed
-
- Anzai A, Choi JL, He S, Fenn AM, Nairz M, Rattik S, McAlpine CS, Mindur JE, Chan CT, Iwamoto Y, Tricot B, Wojtkiewicz GR, Weissleder R, Libby P, Nahrendorf M, Stone JR, Becher B, & Swirski FK. (2017). The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. The Journal of Experimental Medicine, 214(11), 3293–3310. 10.1084/jem.20170689 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical