

Scaffolding protein functional sites using deep learning
- PMID: 35862514
- PMCID: PMC9621694
- DOI: 10.1126/science.abn2100
Scaffolding protein functional sites using deep learning
Abstract
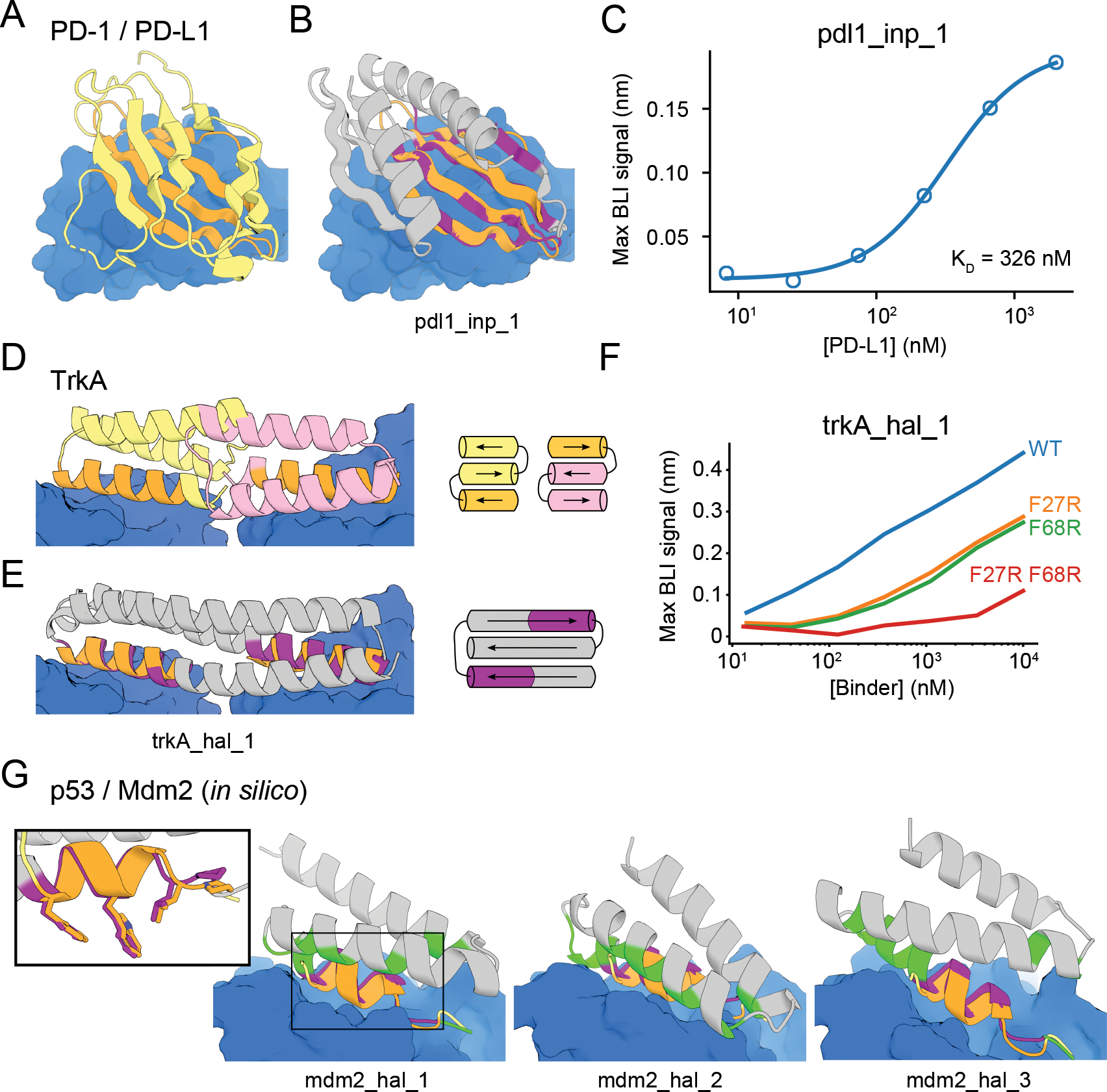
The binding and catalytic functions of proteins are generally mediated by a small number of functional residues held in place by the overall protein structure. Here, we describe deep learning approaches for scaffolding such functional sites without needing to prespecify the fold or secondary structure of the scaffold. The first approach, "constrained hallucination," optimizes sequences such that their predicted structures contain the desired functional site. The second approach, "inpainting," starts from the functional site and fills in additional sequence and structure to create a viable protein scaffold in a single forward pass through a specifically trained RoseTTAFold network. We use these two methods to design candidate immunogens, receptor traps, metalloproteins, enzymes, and protein-binding proteins and validate the designs using a combination of in silico and experimental tests.
Conflict of interest statement
Competing interests
Authors declare that they have no competing interests.
Figures
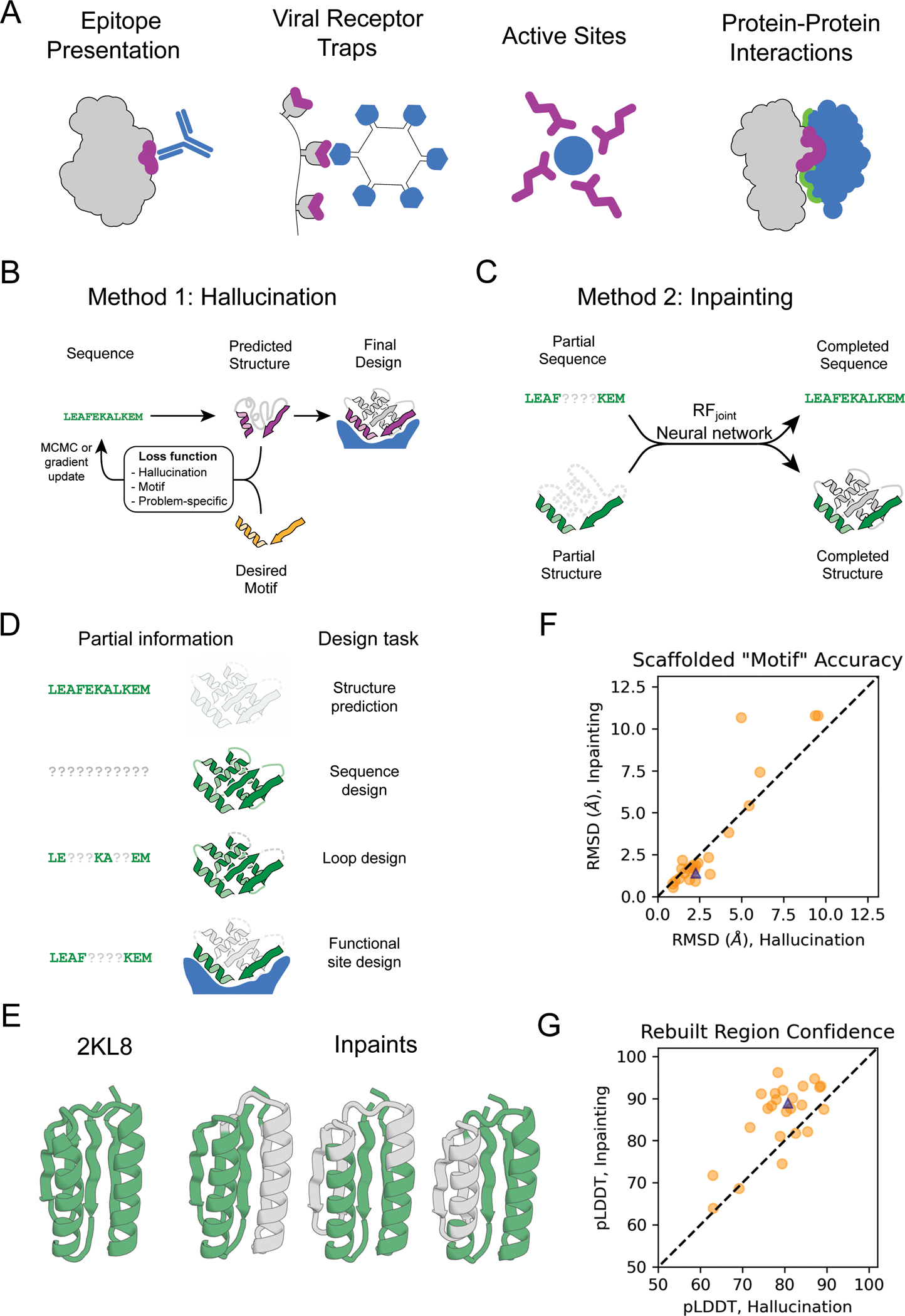
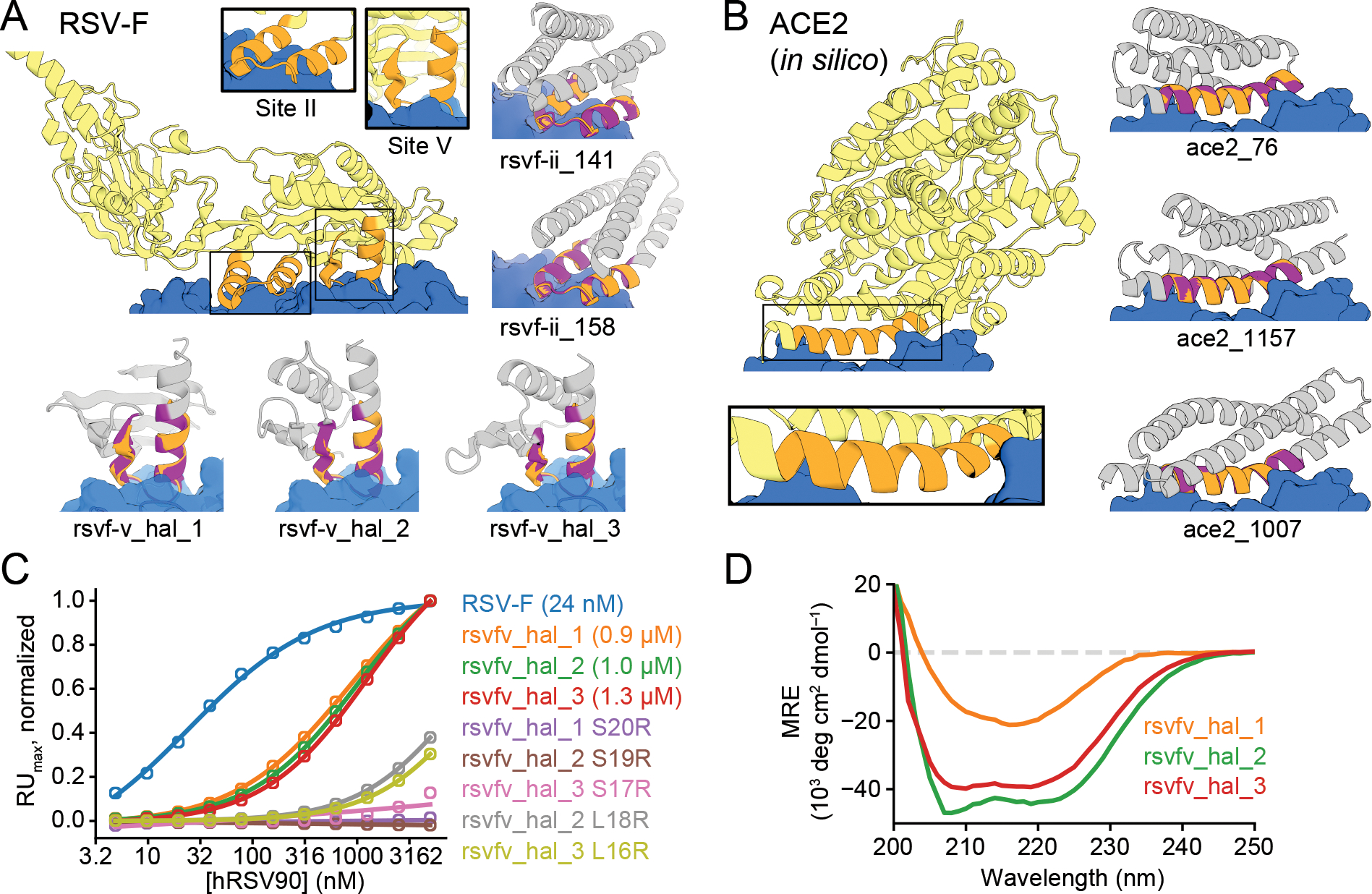
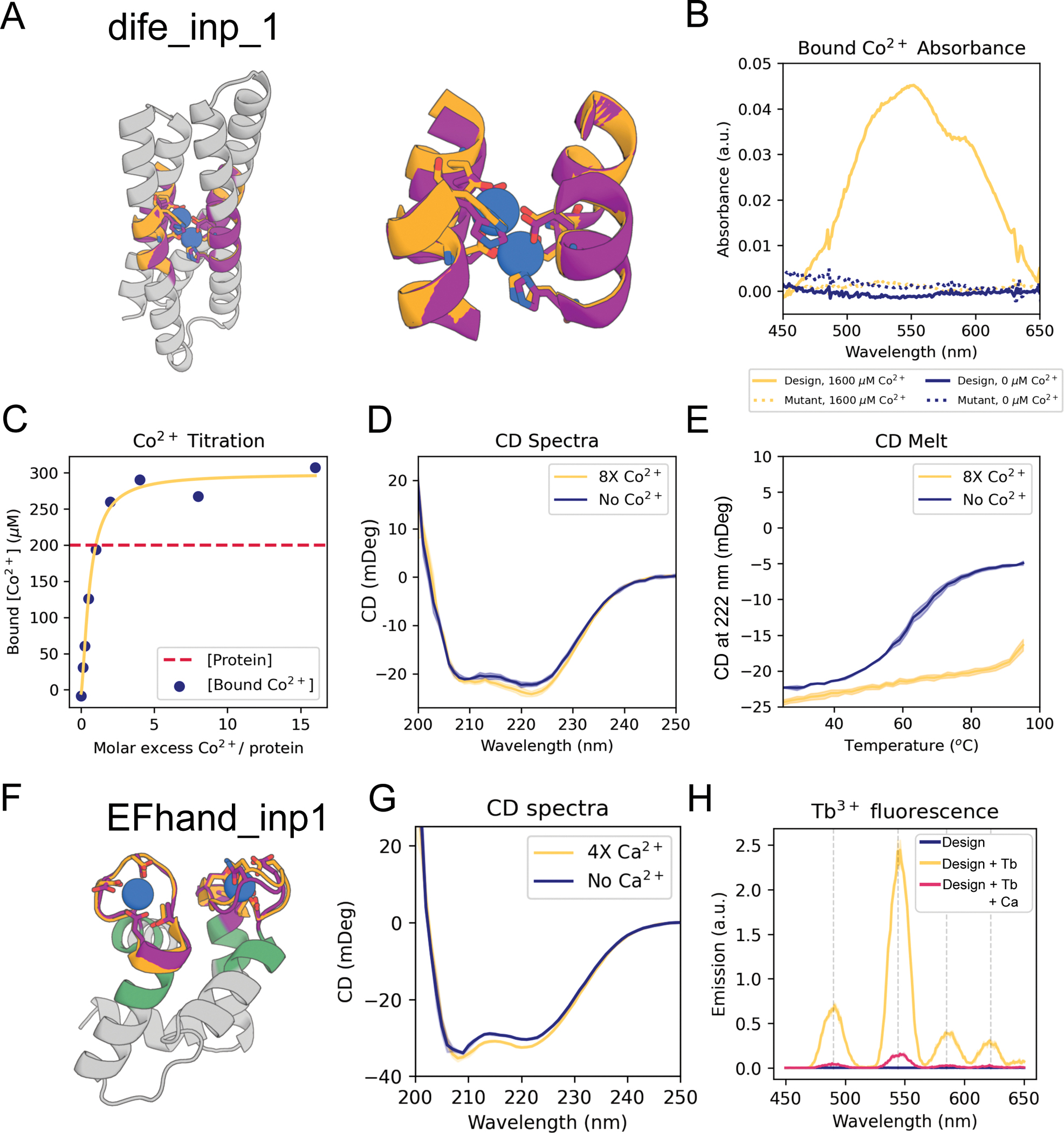
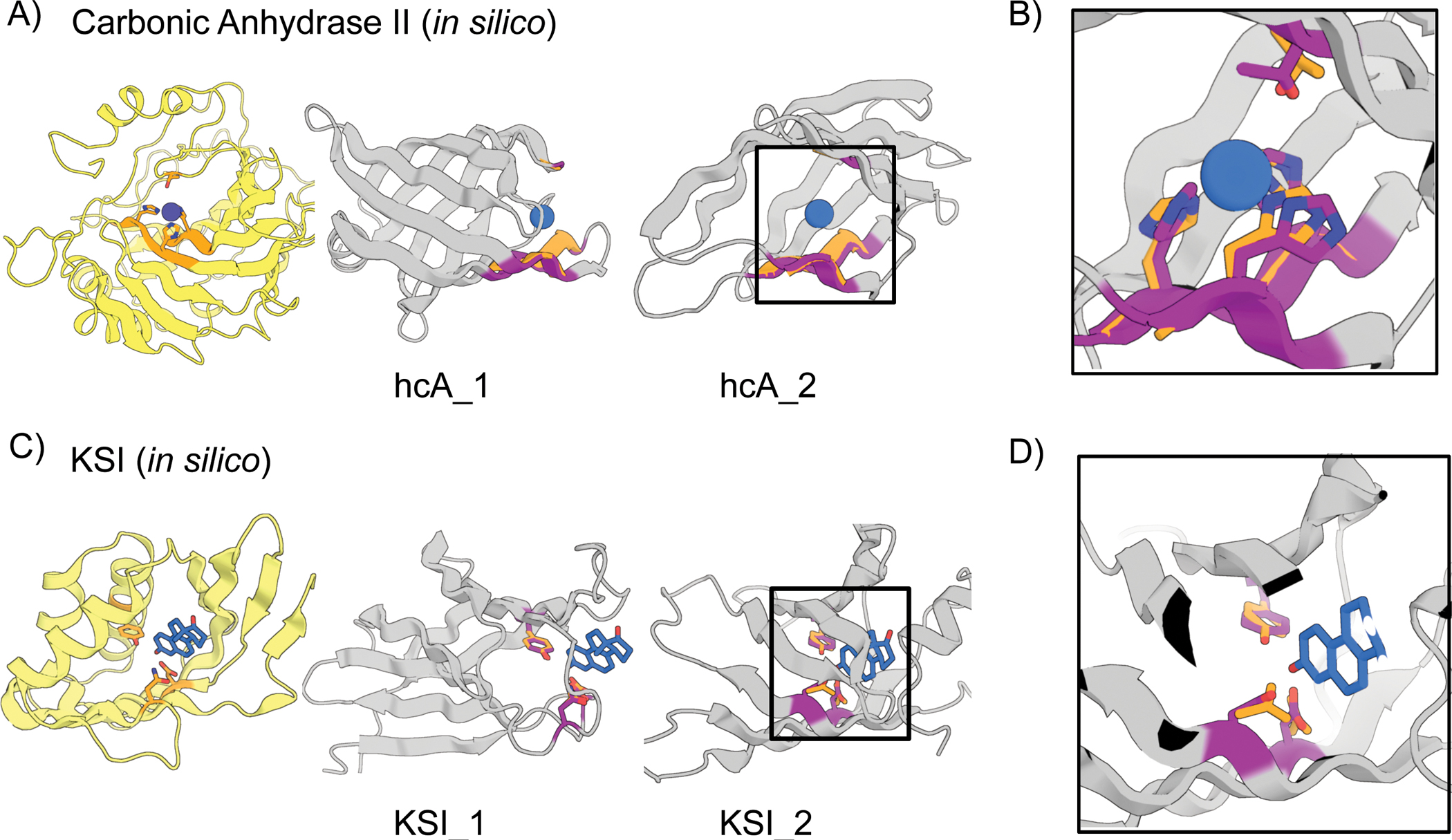
References
-
- Siegel JB, Zanghellini A, Lovick HM, Kiss G, Lambert AR, St. Clair JL, Gallaher J, Hilvert D, Gelb MH, Stoddard BL, Houk KN, Michael FE, Baker D, Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels-Alder Reaction. Science. 329 (2010), doi: 10.1126/science.1190239. - DOI - PMC - PubMed
-
- Cao L, Coventry B, Goreshnik I, Huang B, Park JS, Jude KM, Marković I, Kadam RU, Verschueren KHG, Verstraete K, Walsh STR, Bennett N, Phal A, Yang A, Kozodoy L, DeWitt M, Picton L, Miller L, Strauch E-M, DeBouver ND, Pires A, Bera AK, Halabiya S, Hammerson B, Yang W, Bernard S, Stewart L, Wilson IA, Ruohola-Baker H, Schlessinger J, Lee S, Savvides SN, Garcia KC, Baker D, Design of protein binding proteins from target structure alone. Nature (2022), doi: 10.1038/s41586-022-04654-9. - DOI - PMC - PubMed
-
- Chevalier AA, Silva D, Rocklin GJ, Derrick R, Vergara R, Murapa P, Bernard SM, Zhang L, Yao G, Bahl CD, Miyashita S, Goreshnik I, James T, Bryan M, Fernández-velasco DA, Stewart L, Dong M, Huang X, Massively parallel de novo protein design for targeted therapeutics. Nat. Publ. Group (2017), doi: 10.1038/nature23912. - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
