

Mapping hormone-regulated cell-cell interaction networks in the human breast at single-cell resolution
- PMID: 35863345
- PMCID: PMC9590200
- DOI: 10.1016/j.cels.2022.06.005
Mapping hormone-regulated cell-cell interaction networks in the human breast at single-cell resolution
Abstract
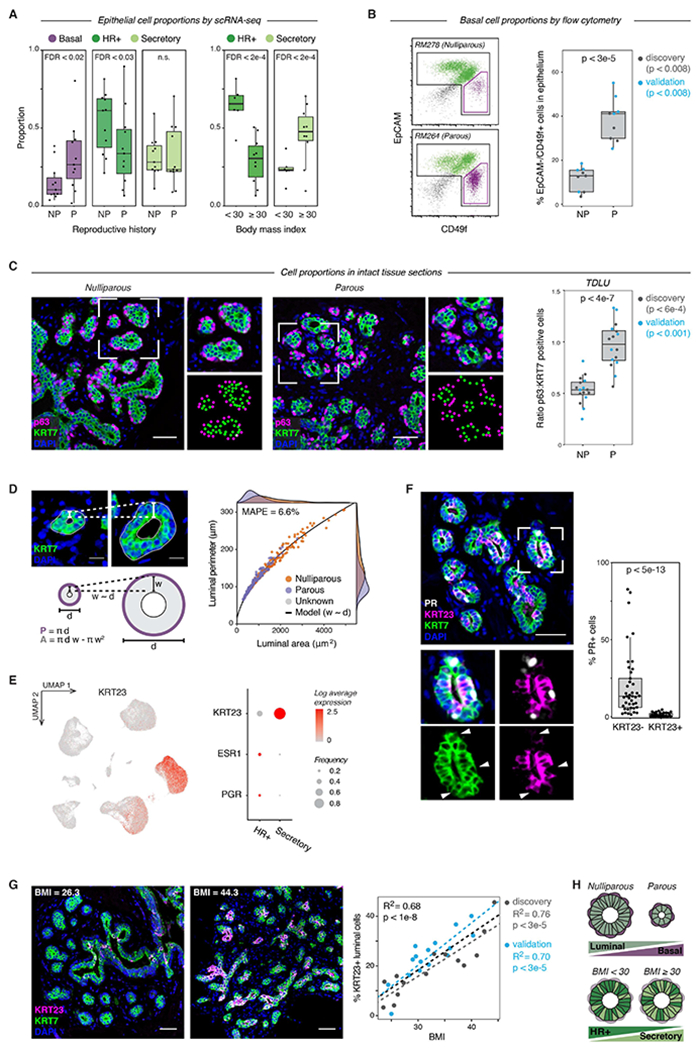
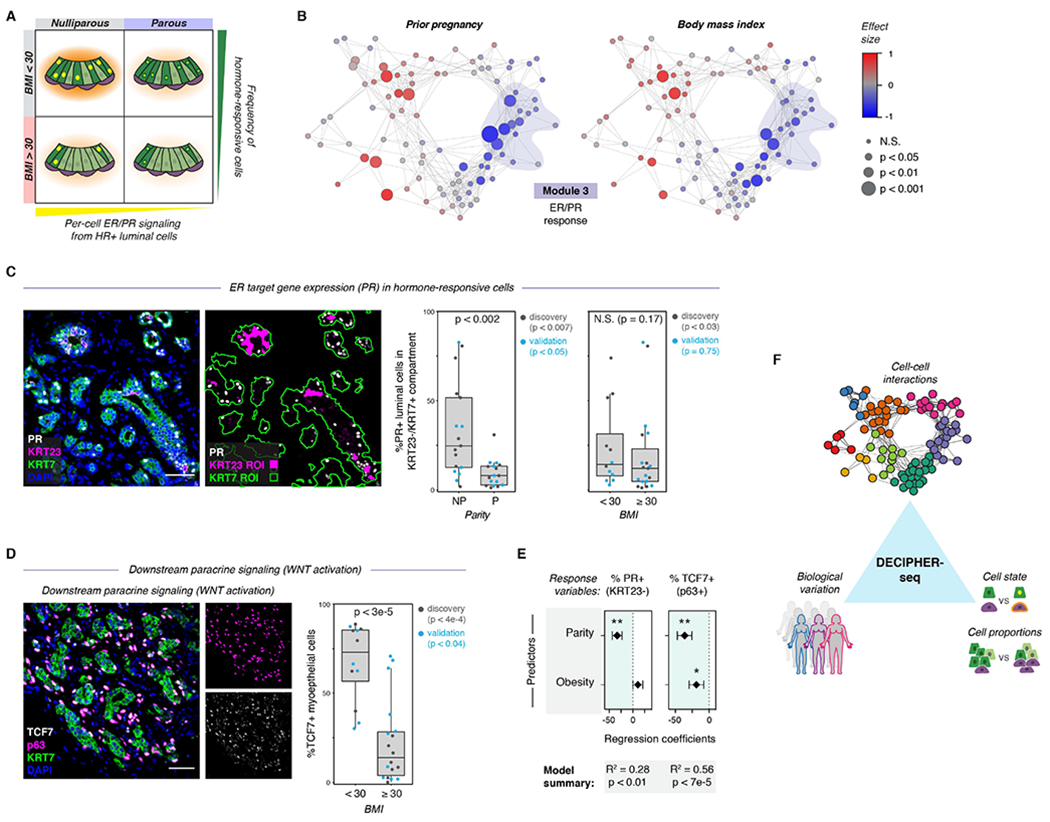
The rise and fall of estrogen and progesterone across menstrual cycles and during pregnancy regulates breast development and modifies cancer risk. How these hormones impact each cell type in the breast remains poorly understood because they act indirectly through paracrine networks. Using single-cell analysis of premenopausal breast tissue, we reveal a network of coordinated transcriptional programs representing the tissue-level response to changing hormone levels. Our computational approach, DECIPHER-seq, leverages person-to-person variability in breast composition and cell state to uncover programs that co-vary across individuals. We use differences in cell-type proportions to infer a subset of programs that arise from direct cell-cell interactions regulated by hormones. Further, we demonstrate that prior pregnancy and obesity modify hormone responsiveness through distinct mechanisms: obesity reduces the proportion of hormone-responsive cells, whereas pregnancy dampens the direct response of these cells to hormones. Together, these results provide a comprehensive map of the cycling human breast.
Keywords: cell-cell interactions; hormone signaling; human breast; sample heterogeneity; scRNA-seq; single-cell genomics.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests Z.J.G. and C.S.M. hold patents related to the MULTI-seq barcoding method. Z.J.G. is an equity holder in Scribe Biosciences and Provenance bio and a member of the scientific advisory board of Serotiny Bio. C.S.M. is a consultant for ImYoo. Since January 10, 2022, L.M.M. is an employee of Genentech, a member of the Roche group.
Figures

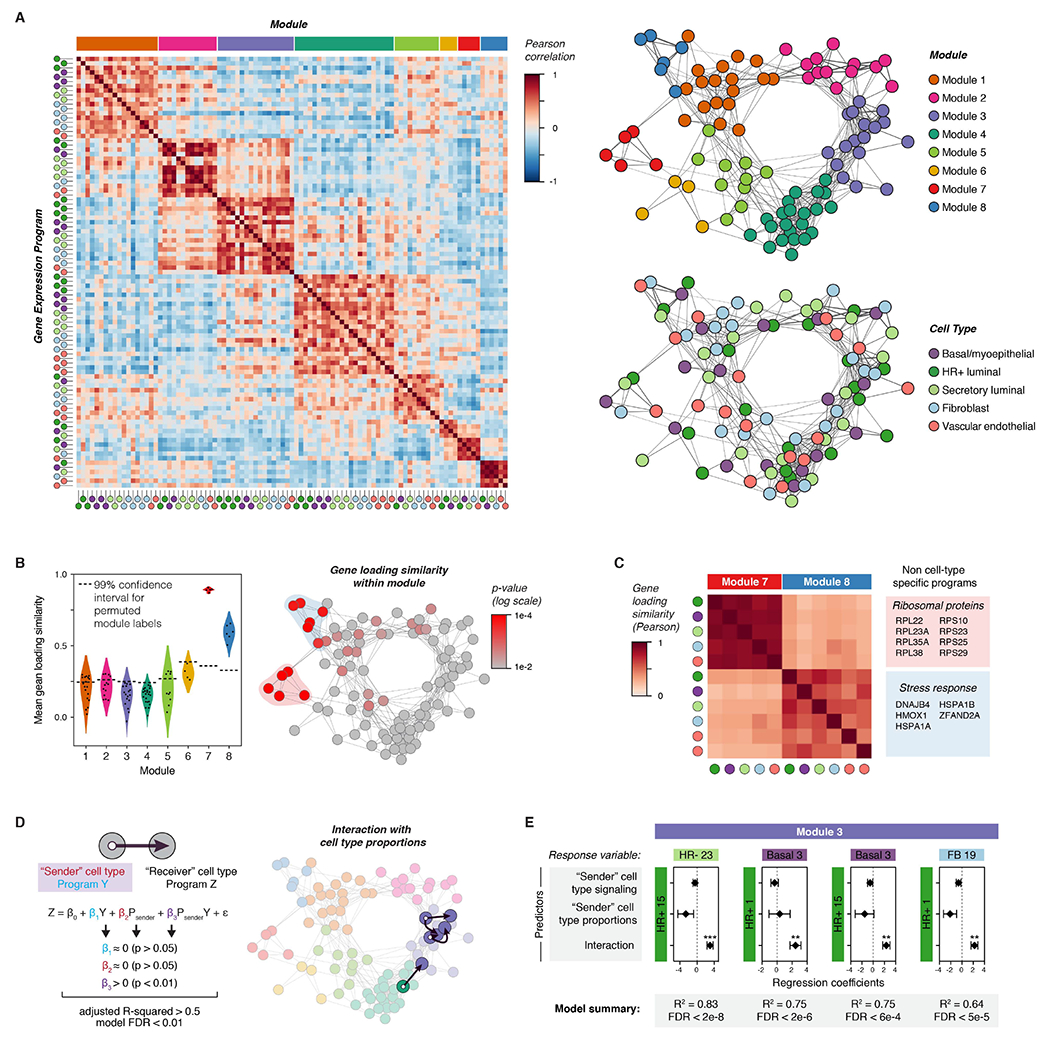
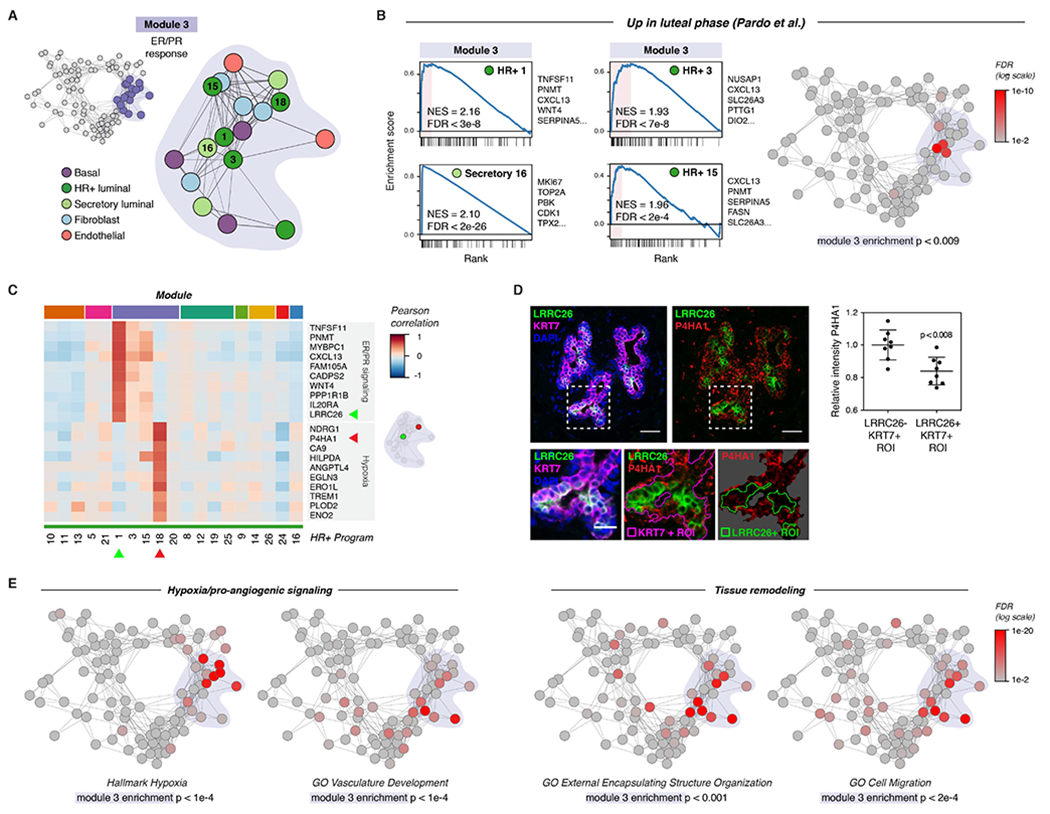
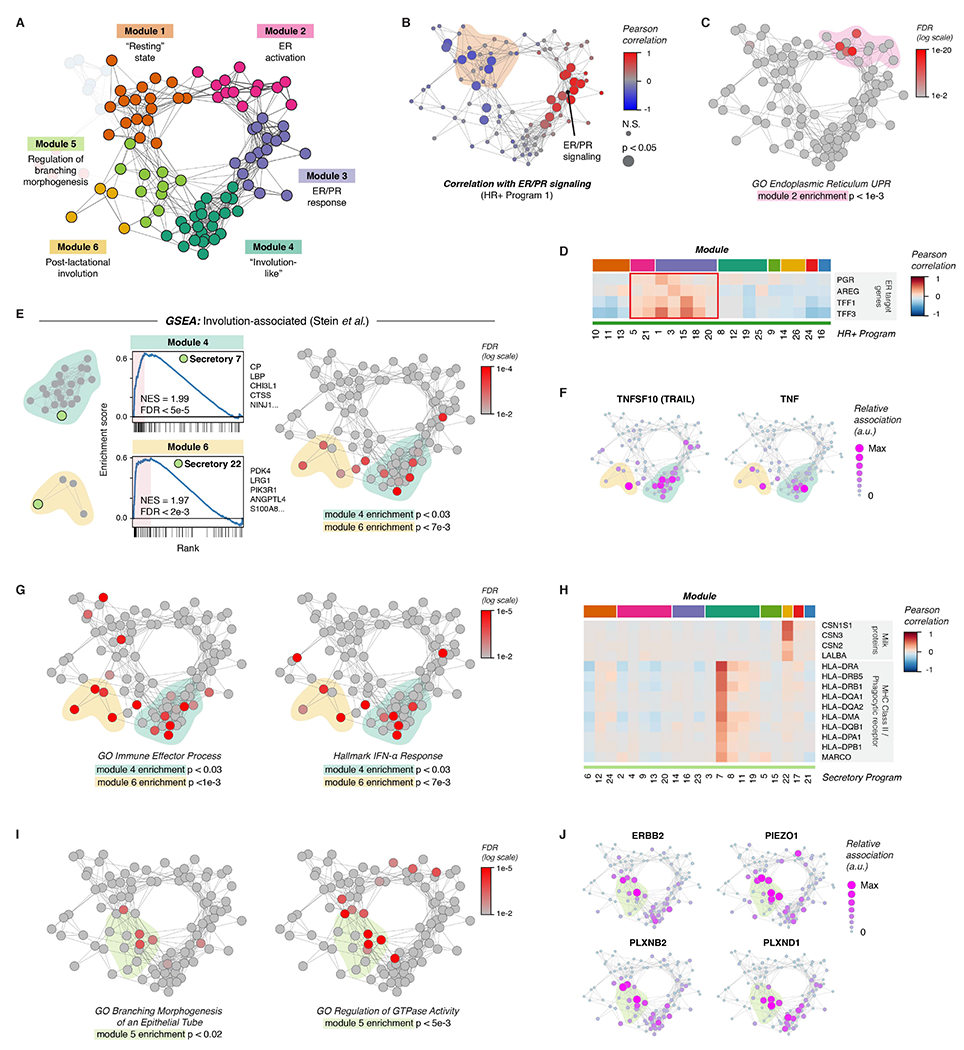
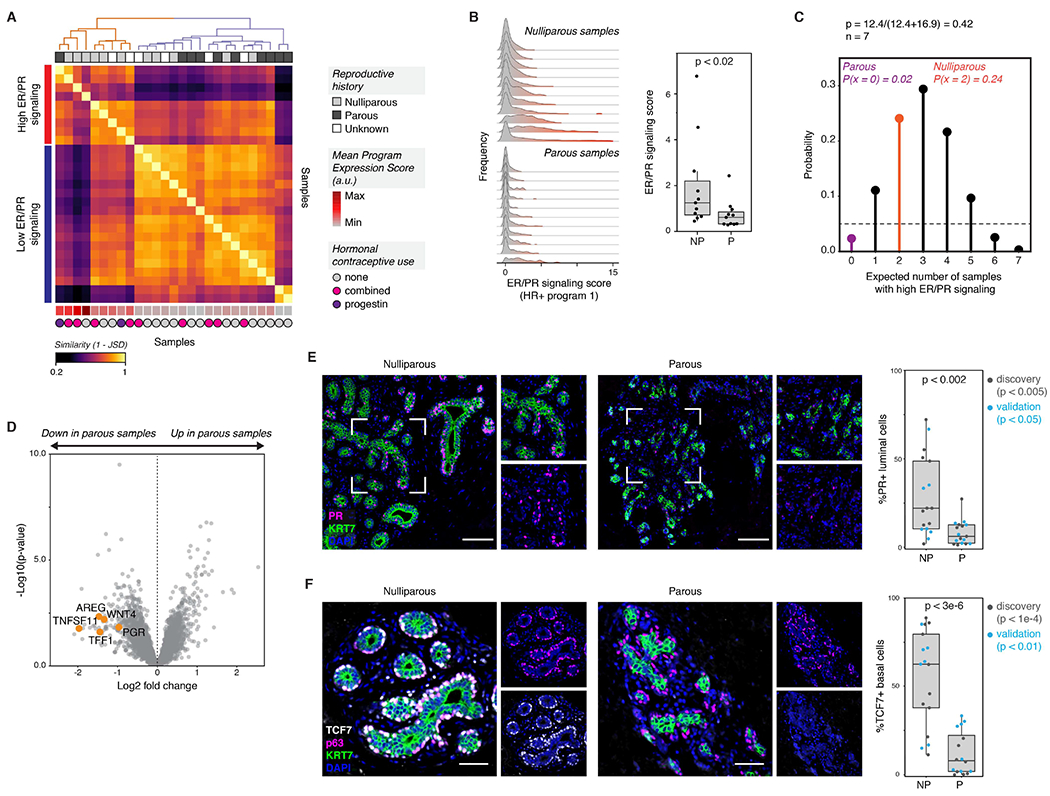
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
