

"Stripe" transcription factors provide accessibility to co-binding partners in mammalian genomes
- PMID: 35863348
- PMCID: PMC9481673
- DOI: 10.1016/j.molcel.2022.06.029
"Stripe" transcription factors provide accessibility to co-binding partners in mammalian genomes
Abstract
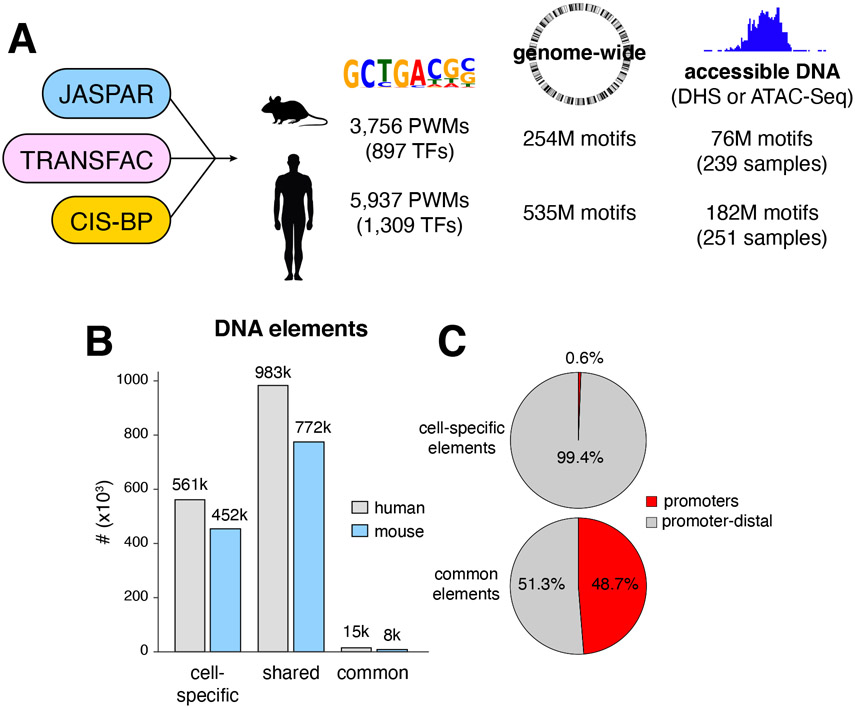
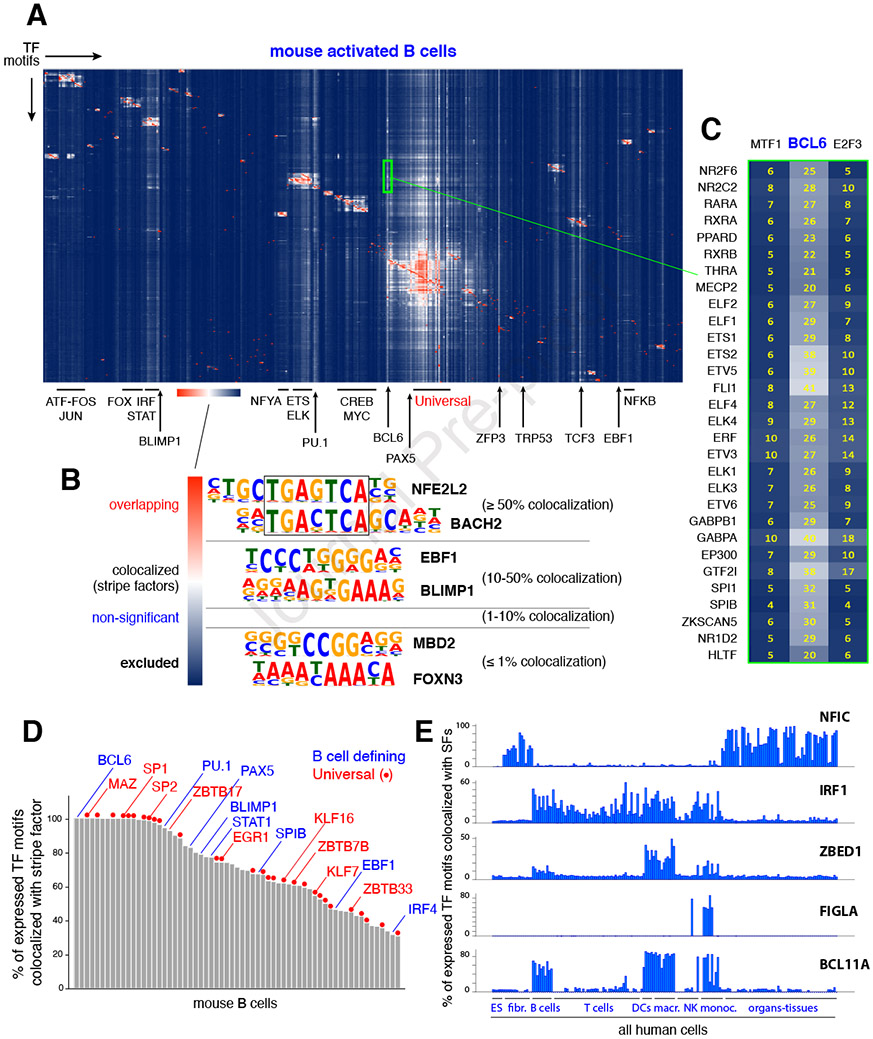
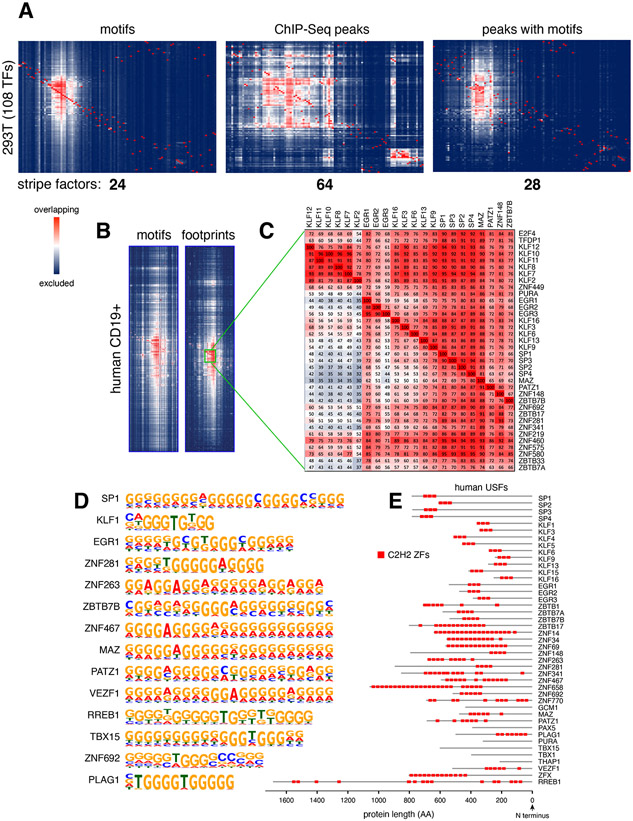
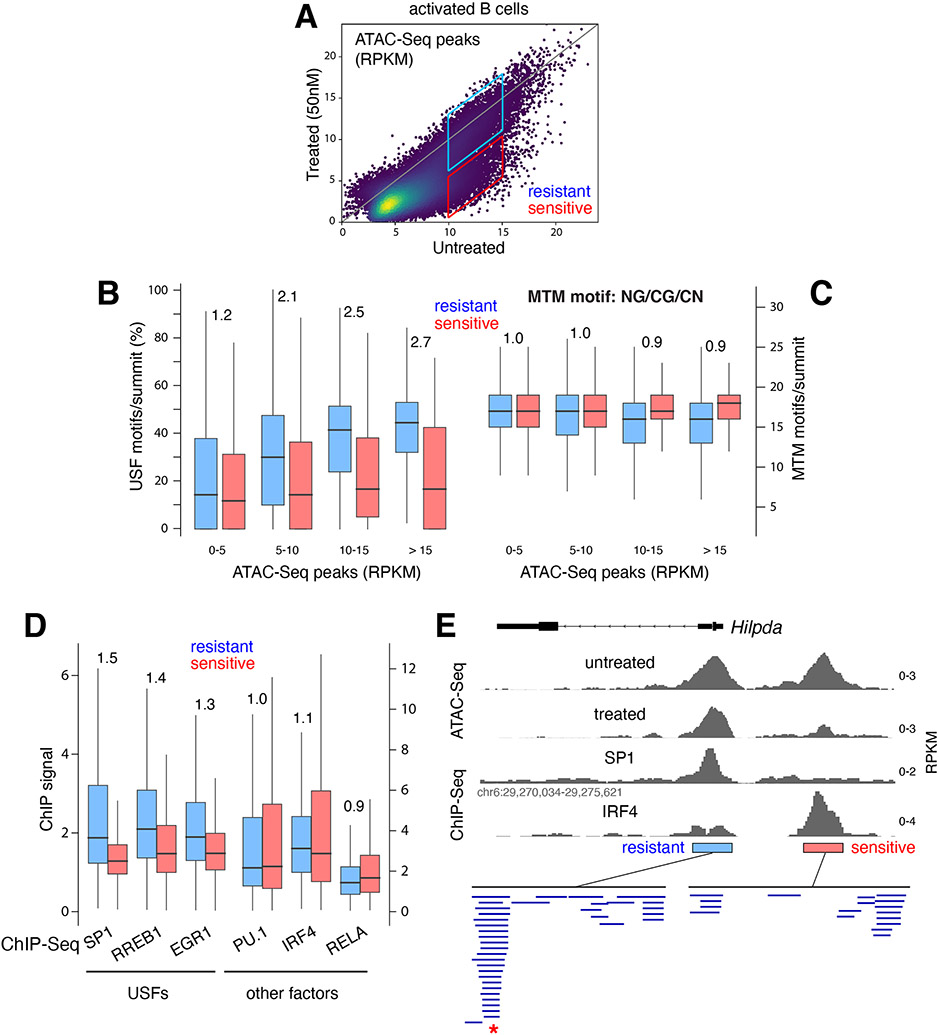
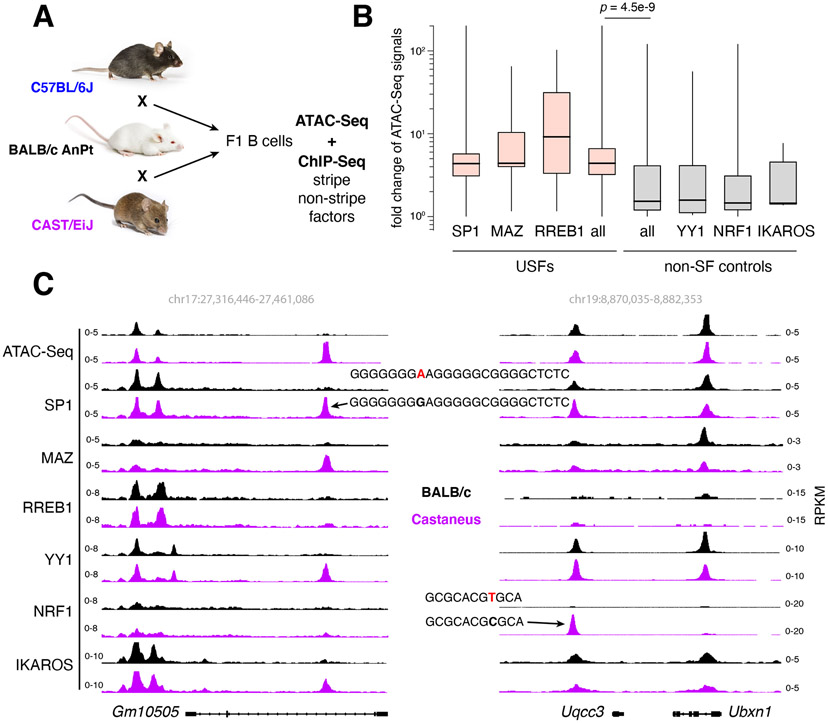
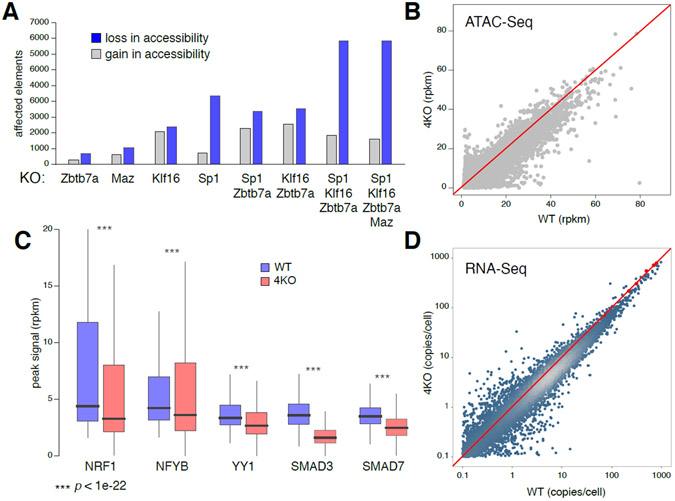
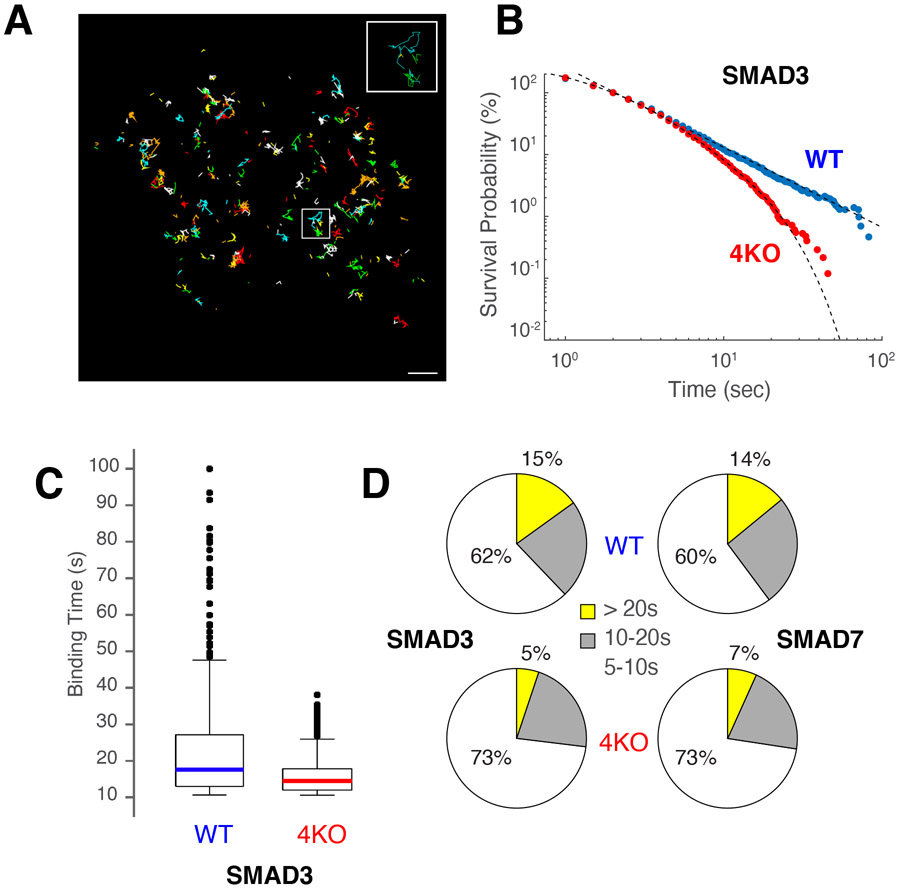
Regulatory elements activate promoters by recruiting transcription factors (TFs) to specific motifs. Notably, TF-DNA interactions often depend on cooperativity with colocalized partners, suggesting an underlying cis-regulatory syntax. To explore TF cooperativity in mammals, we analyze ∼500 mouse and human primary cells by combining an atlas of TF motifs, footprints, ChIP-seq, transcriptomes, and accessibility. We uncover two TF groups that colocalize with most expressed factors, forming stripes in hierarchical clustering maps. The first group includes lineage-determining factors that occupy DNA elements broadly, consistent with their key role in tissue-specific transcription. The second one, dubbed universal stripe factors (USFs), comprises ∼30 SP, KLF, EGR, and ZBTB family members that recognize overlapping GC-rich sequences in all tissues analyzed. Knockouts and single-molecule tracking reveal that USFs impart accessibility to colocalized partners and increase their residence time. Mammalian cells have thus evolved a TF superfamily with overlapping DNA binding that facilitate chromatin accessibility.
Keywords: DNA motifs; chromatin accessibility; enhancer syntax; gene expression; mammalian genomes; regulatory elements; single molecule tracking; transcription factors.
Copyright © 2022 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests S.P. and L.V. are employees of Astra Zeneca and may own stock or stock options.
Figures
References
-
- Bassuk AG, and Leiden JM (1995). A direct physical association between ETS and AP-1 transcription factors in normal human T cells. Immunity 3, 223–237. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
