

Hijacking of transcriptional condensates by endogenous retroviruses
- PMID: 35864192
- PMCID: PMC9355880
- DOI: 10.1038/s41588-022-01132-w
Hijacking of transcriptional condensates by endogenous retroviruses
Abstract
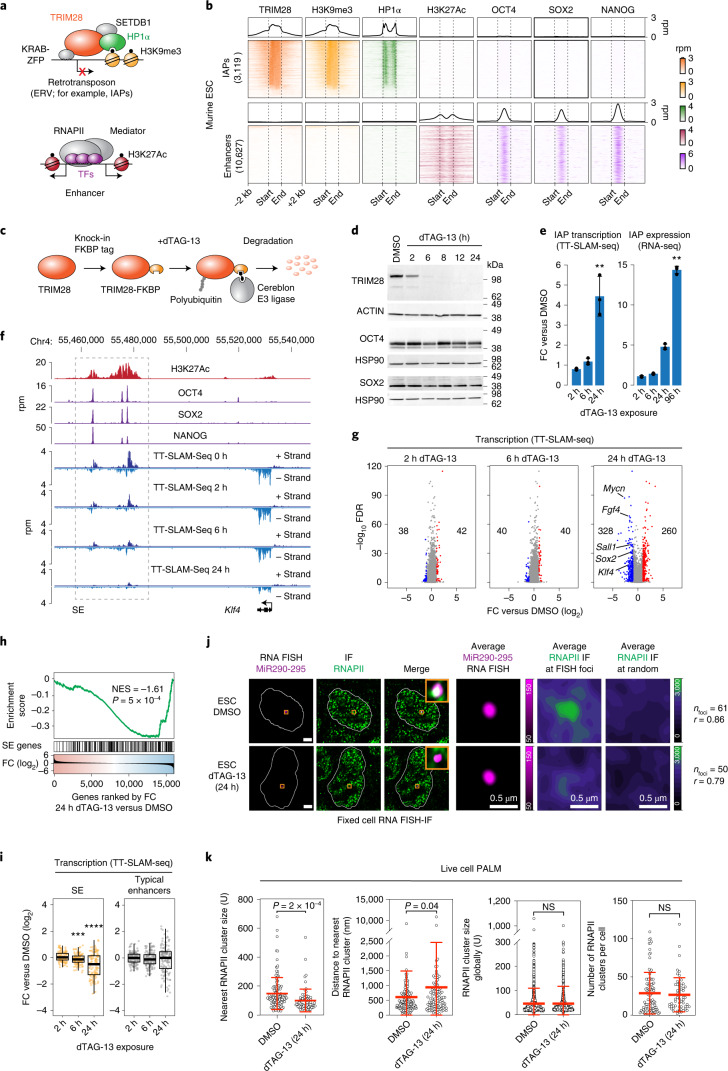
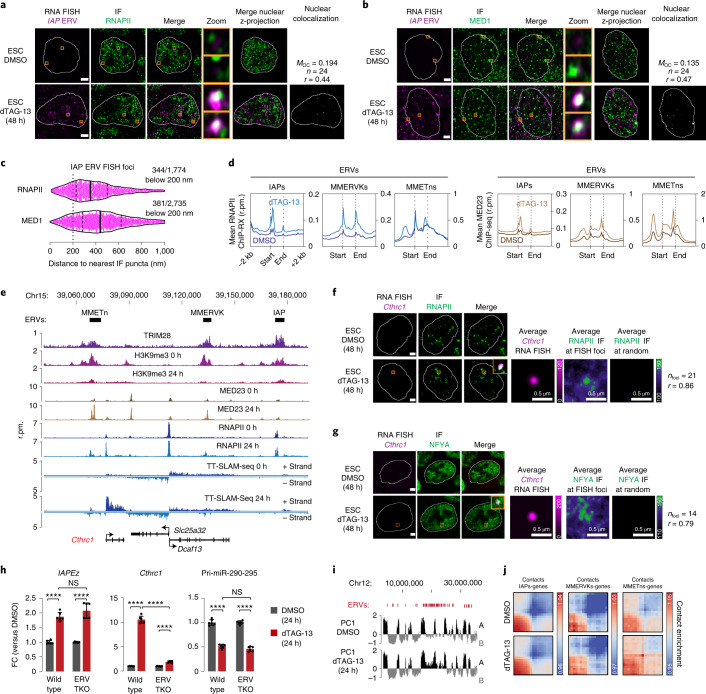
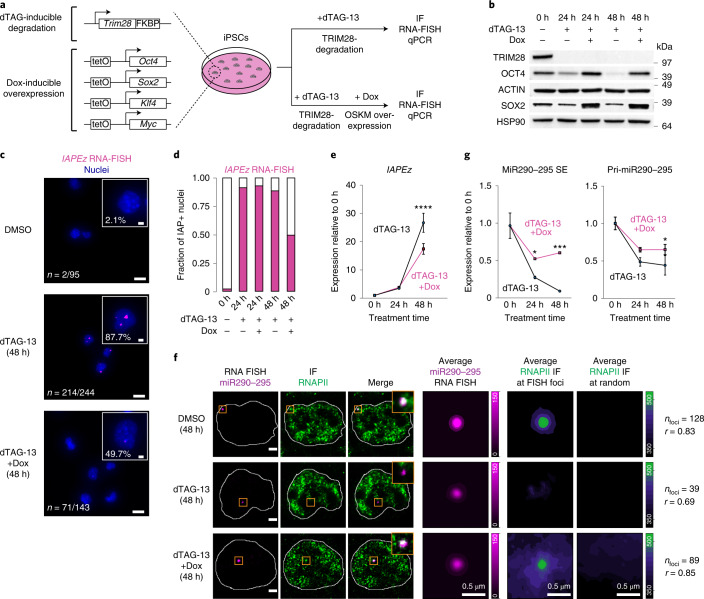
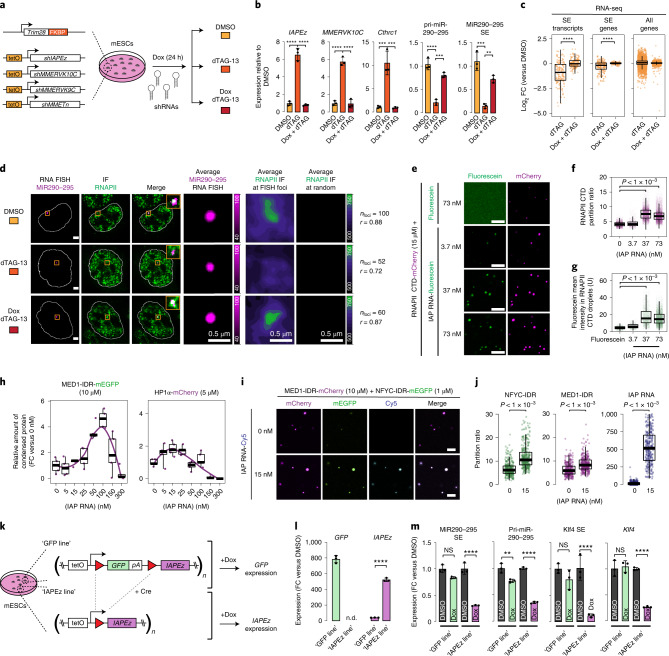
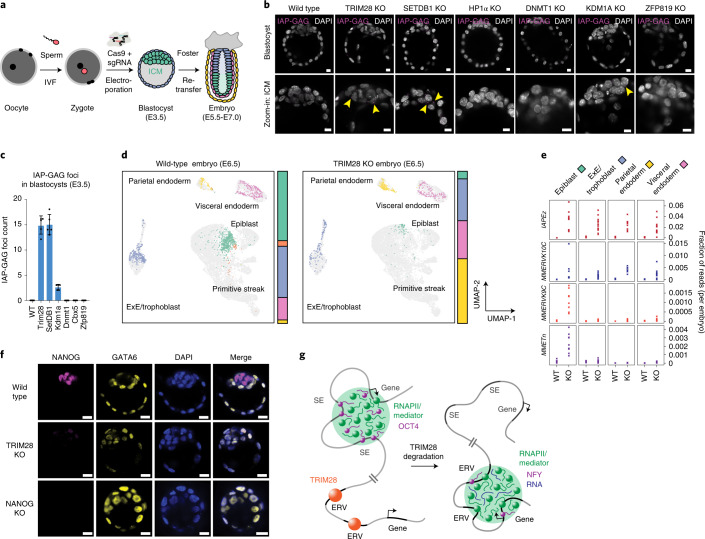
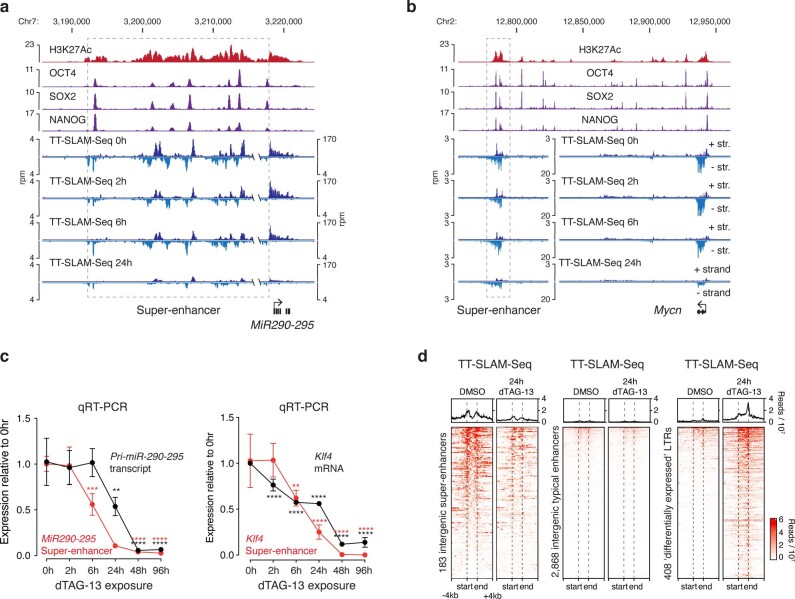
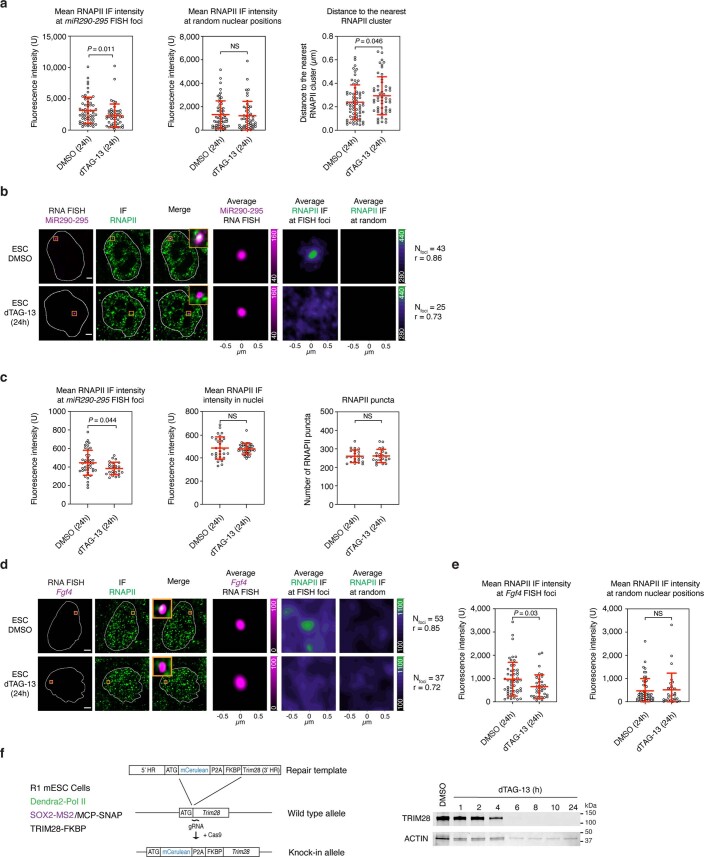
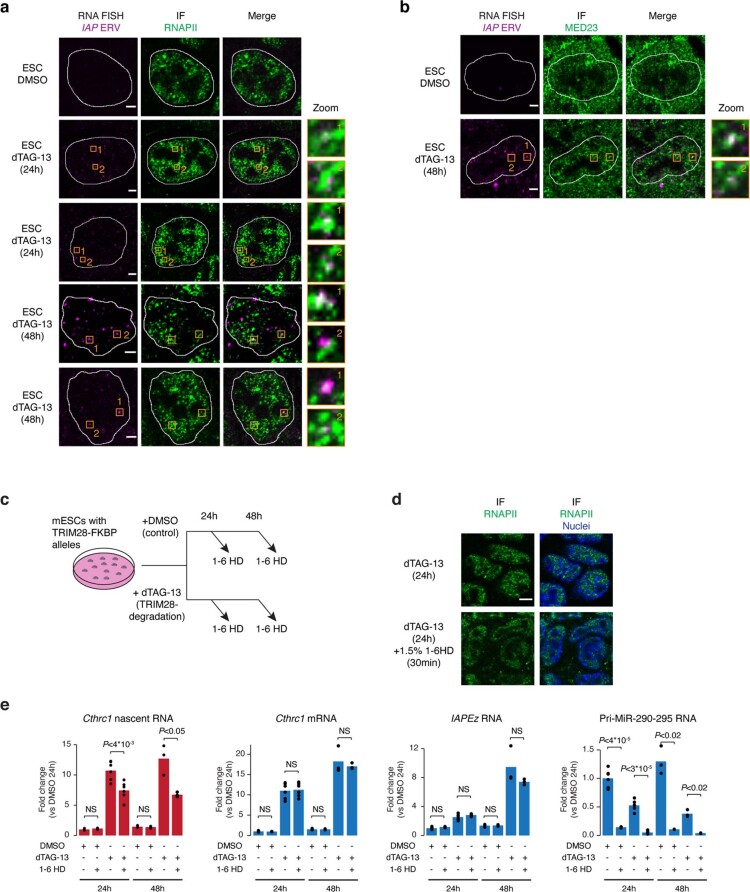
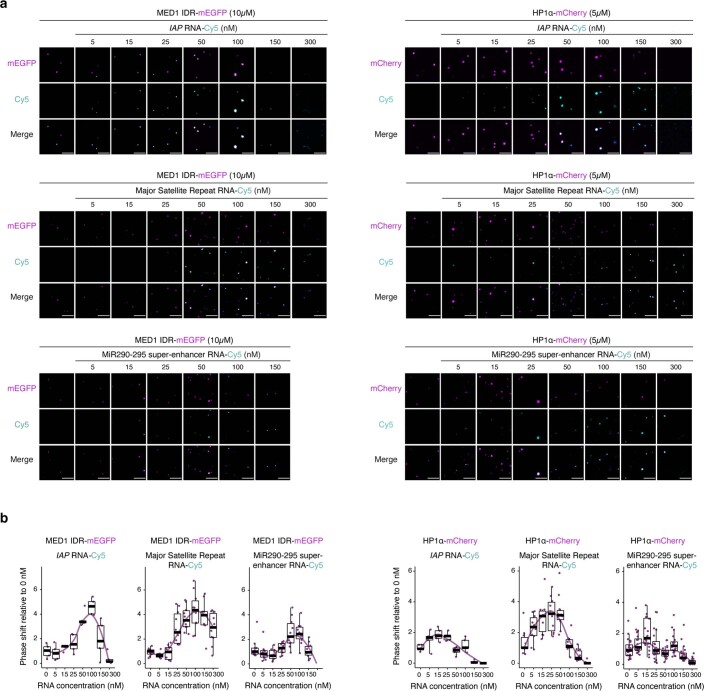
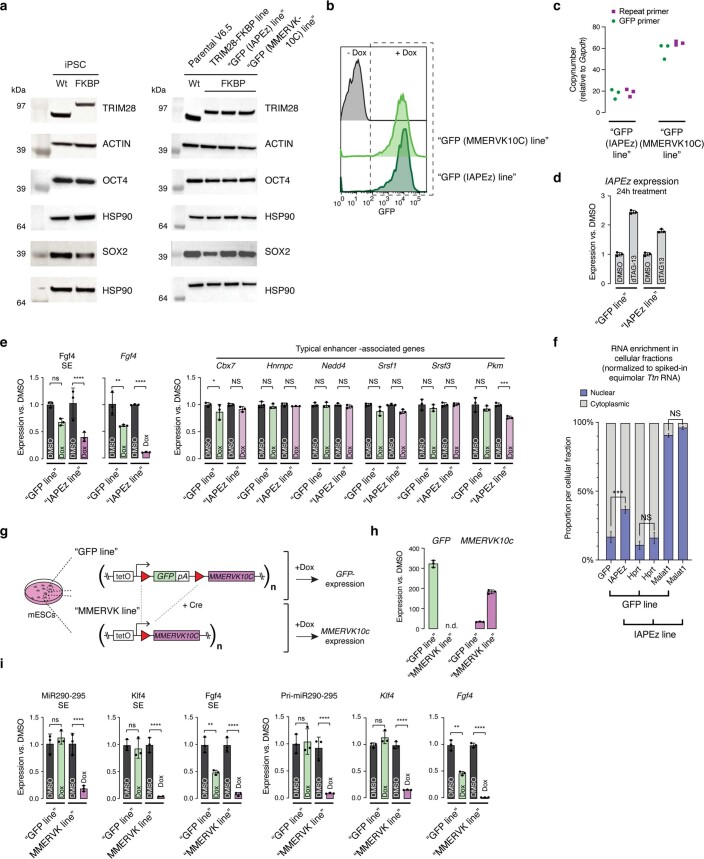
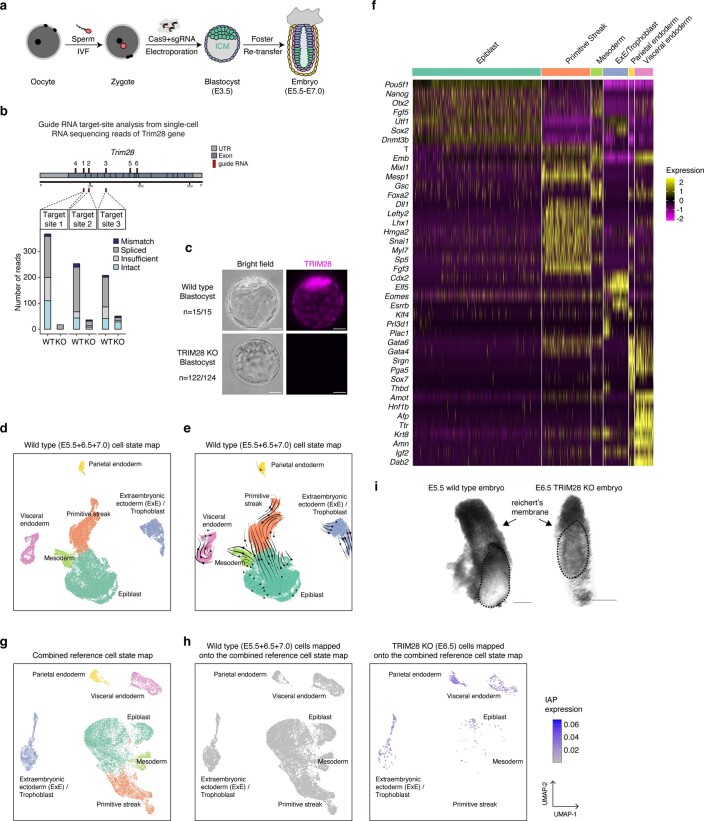
Most endogenous retroviruses (ERVs) in mammals are incapable of retrotransposition; therefore, why ERV derepression is associated with lethality during early development has been a mystery. Here, we report that rapid and selective degradation of the heterochromatin adapter protein TRIM28 triggers dissociation of transcriptional condensates from loci encoding super-enhancer (SE)-driven pluripotency genes and their association with transcribed ERV loci in murine embryonic stem cells. Knockdown of ERV RNAs or forced expression of SE-enriched transcription factors rescued condensate localization at SEs in TRIM28-degraded cells. In a biochemical reconstitution system, ERV RNA facilitated partitioning of RNA polymerase II and the Mediator coactivator into phase-separated droplets. In TRIM28 knockout mouse embryos, single-cell RNA-seq analysis revealed specific depletion of pluripotent lineages. We propose that coding and noncoding nascent RNAs, including those produced by retrotransposons, may facilitate 'hijacking' of transcriptional condensates in various developmental and disease contexts.
© 2022. The Author(s).
Conflict of interest statement
S.L.A. declares competing interest based on a granted patent related to SLAMseq. S.L.A. is co-founder, advisor and member of the board of QUANTRO Therapeutics GmbH
Figures
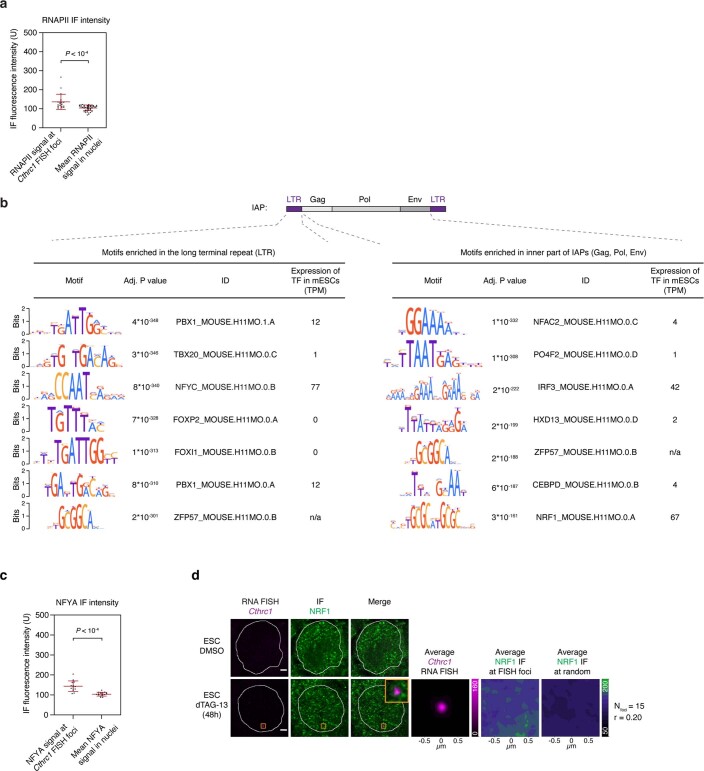
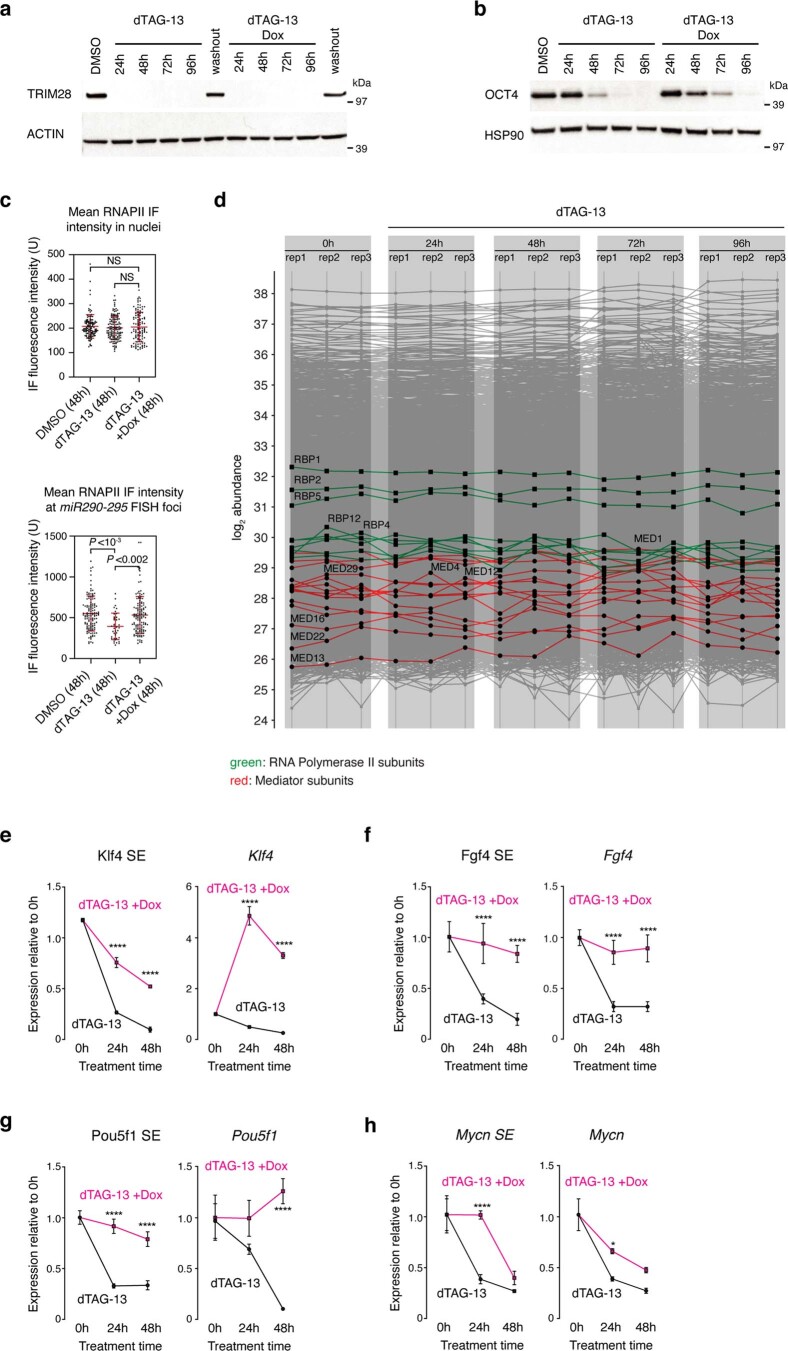
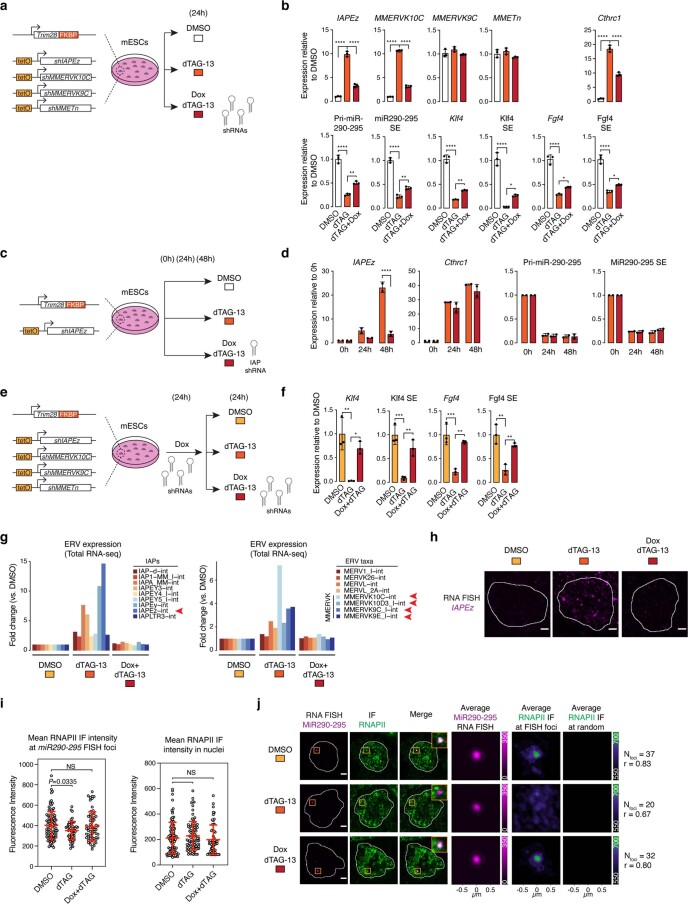
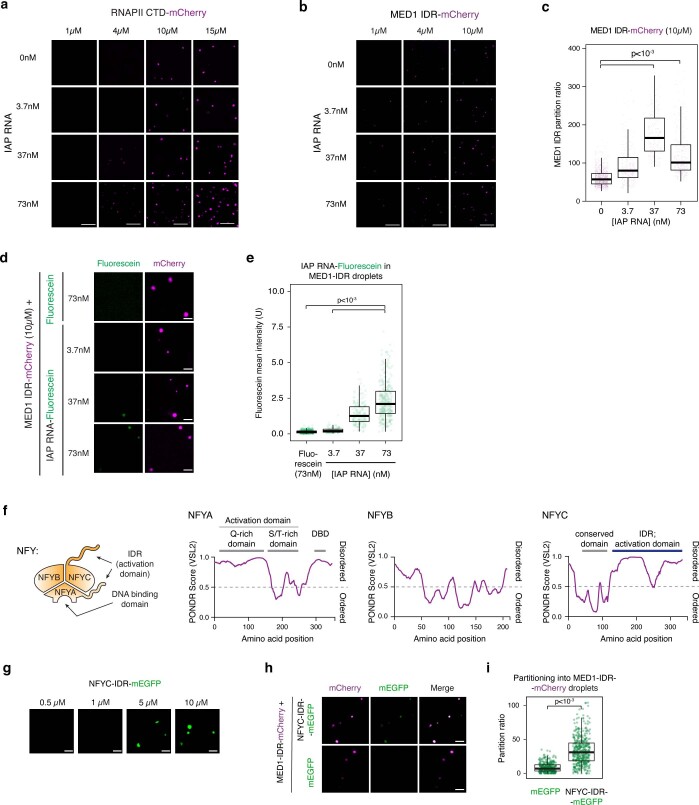
Comment in
-
Endogenous retroviruses steer transcriptional condensates away from pluripotency.Nat Genet. 2022 Aug;54(8):1068-1069. doi: 10.1038/s41588-022-01111-1. Nat Genet. 2022. PMID: 35864191 No abstract available.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
