

Systematic discovery of biomolecular condensate-specific protein phosphorylation
- PMID: 35864335
- PMCID: PMC9512703
- DOI: 10.1038/s41589-022-01062-y
Systematic discovery of biomolecular condensate-specific protein phosphorylation
Abstract
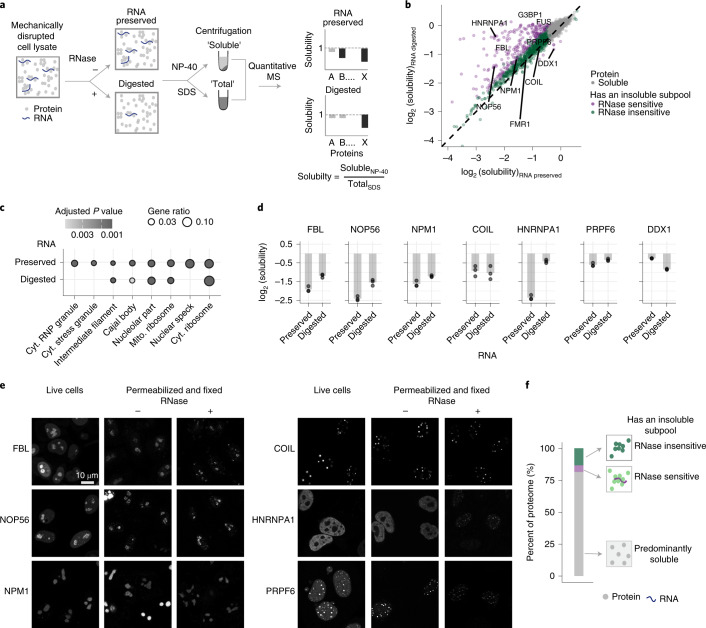
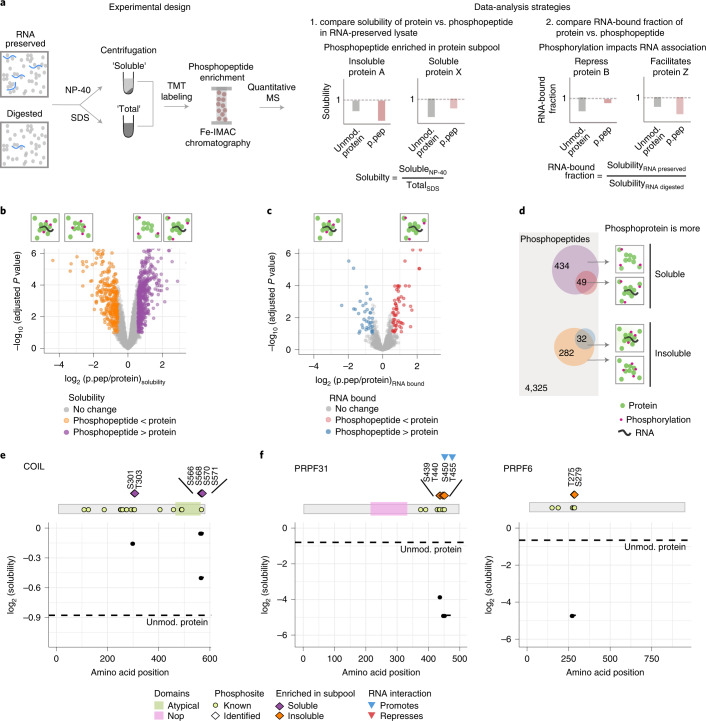
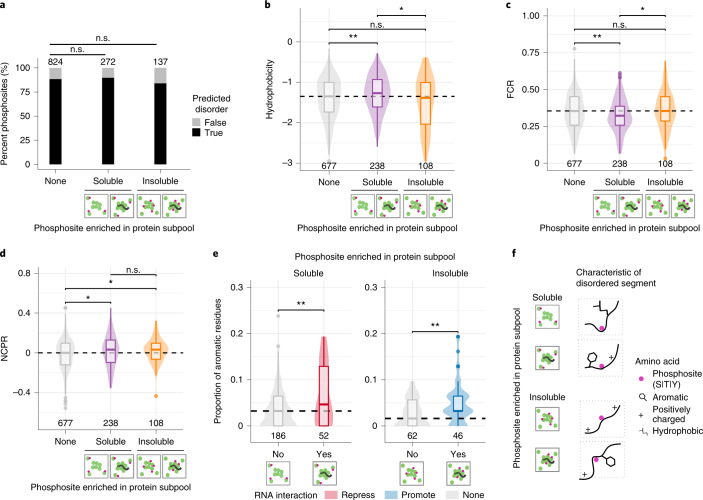
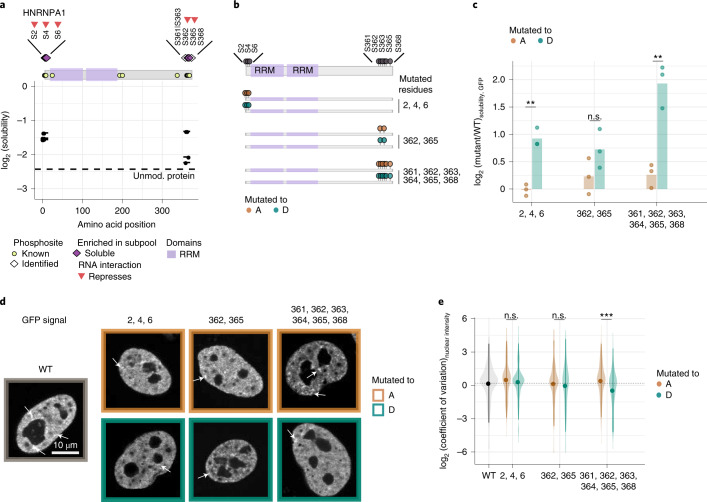
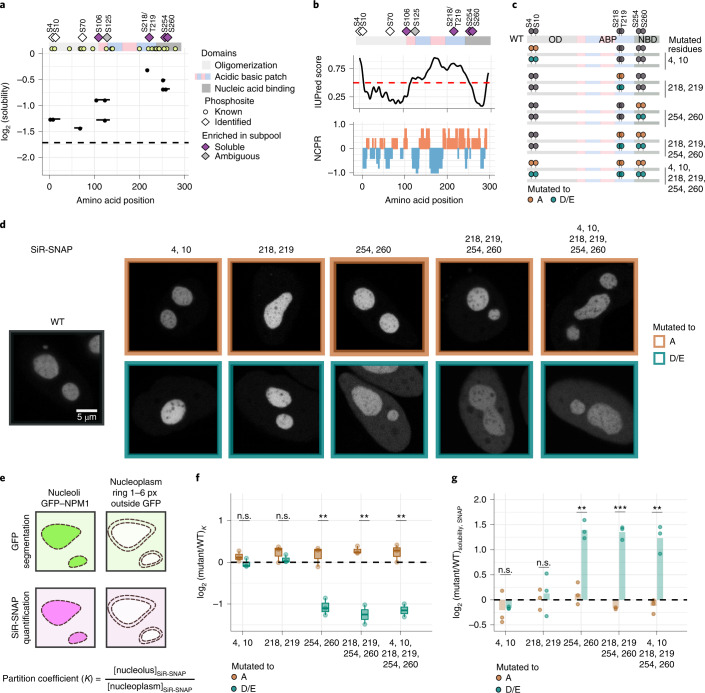
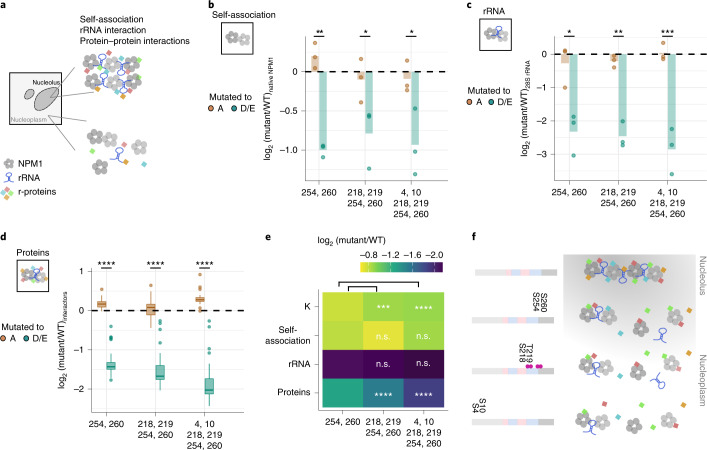
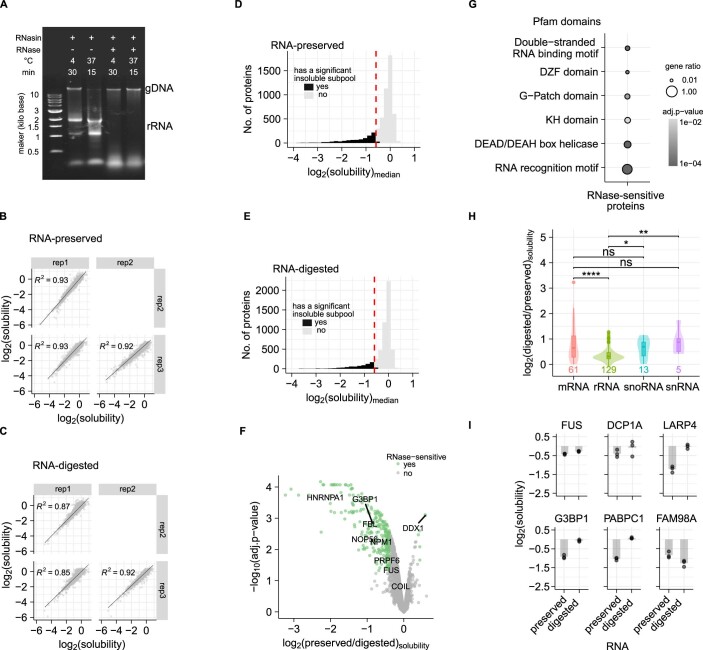
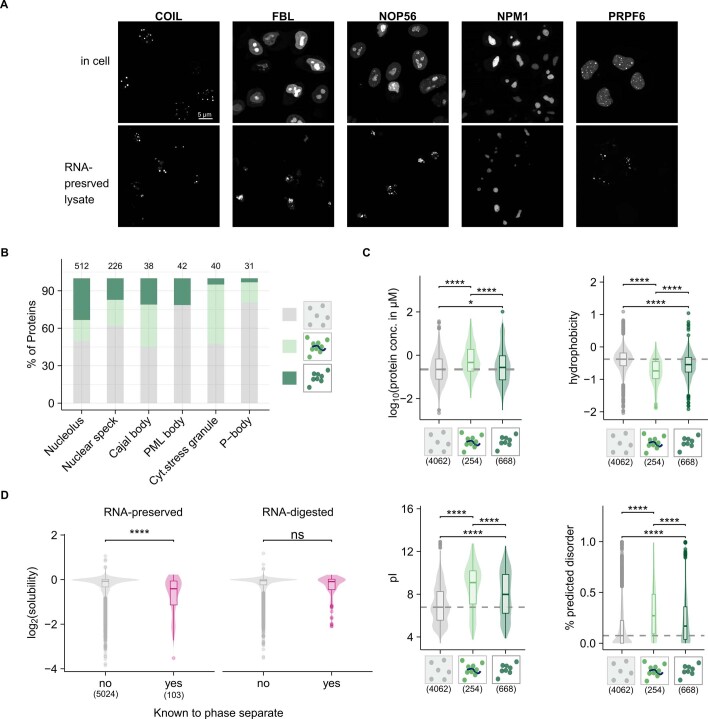
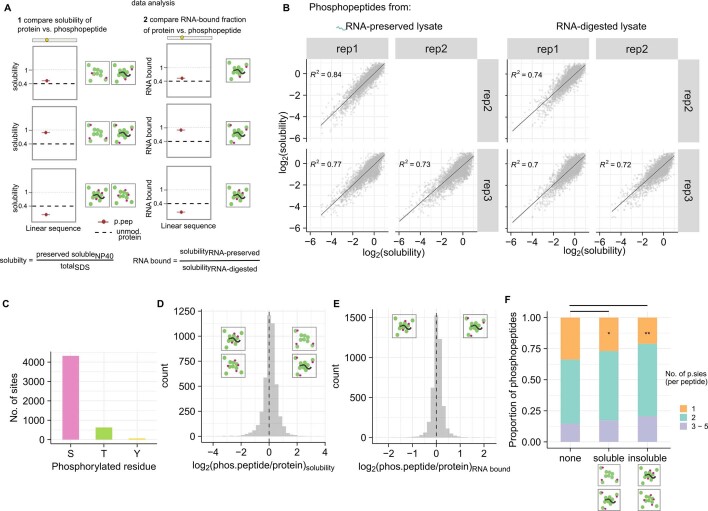
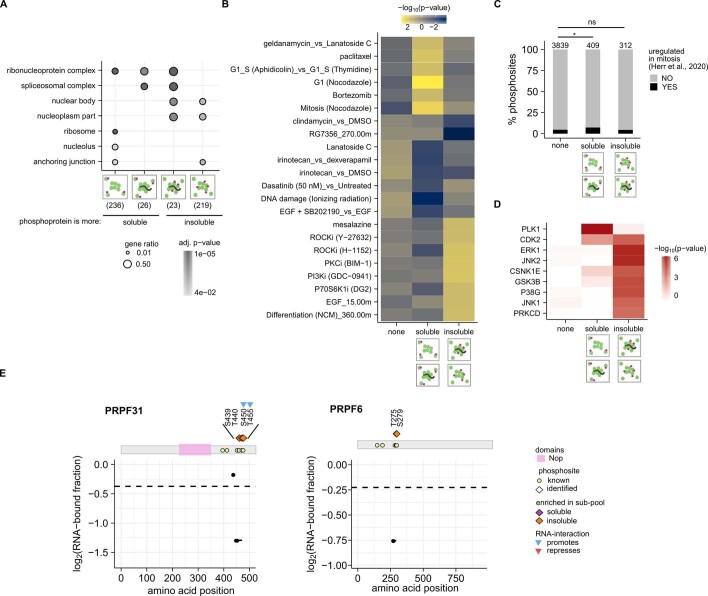
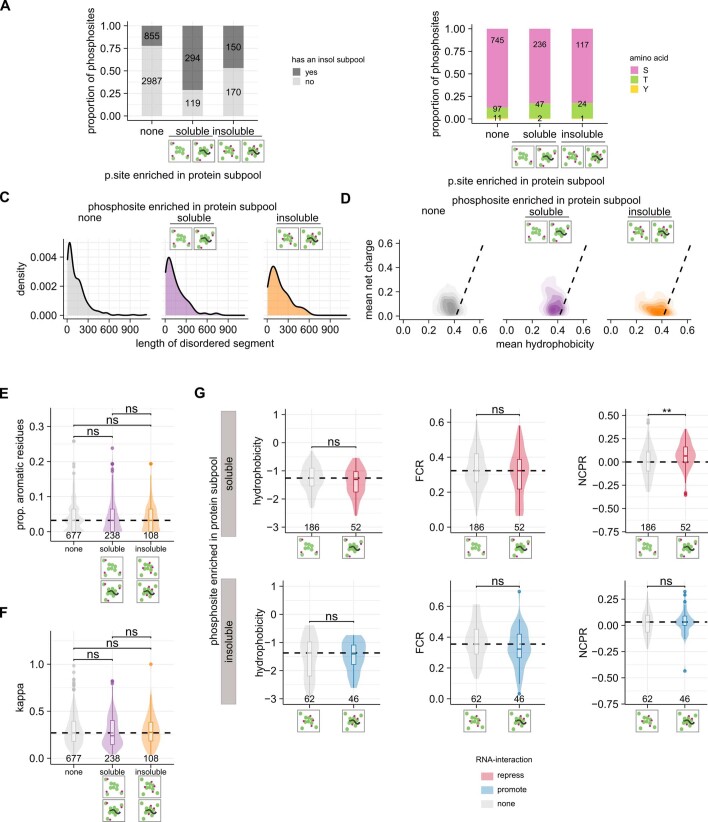
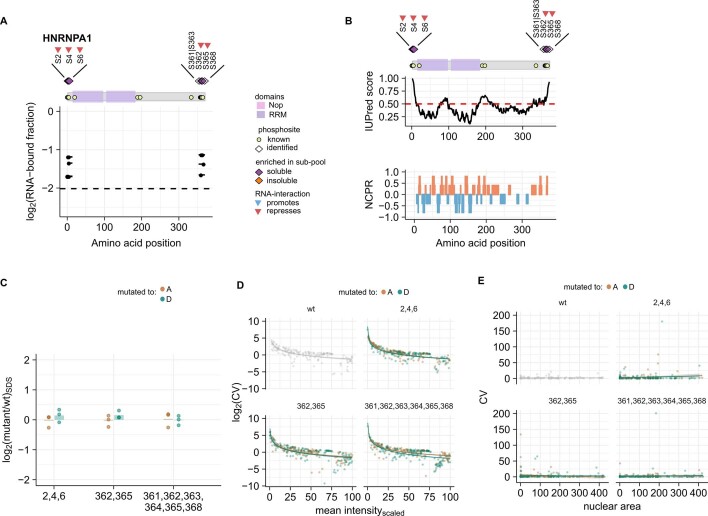
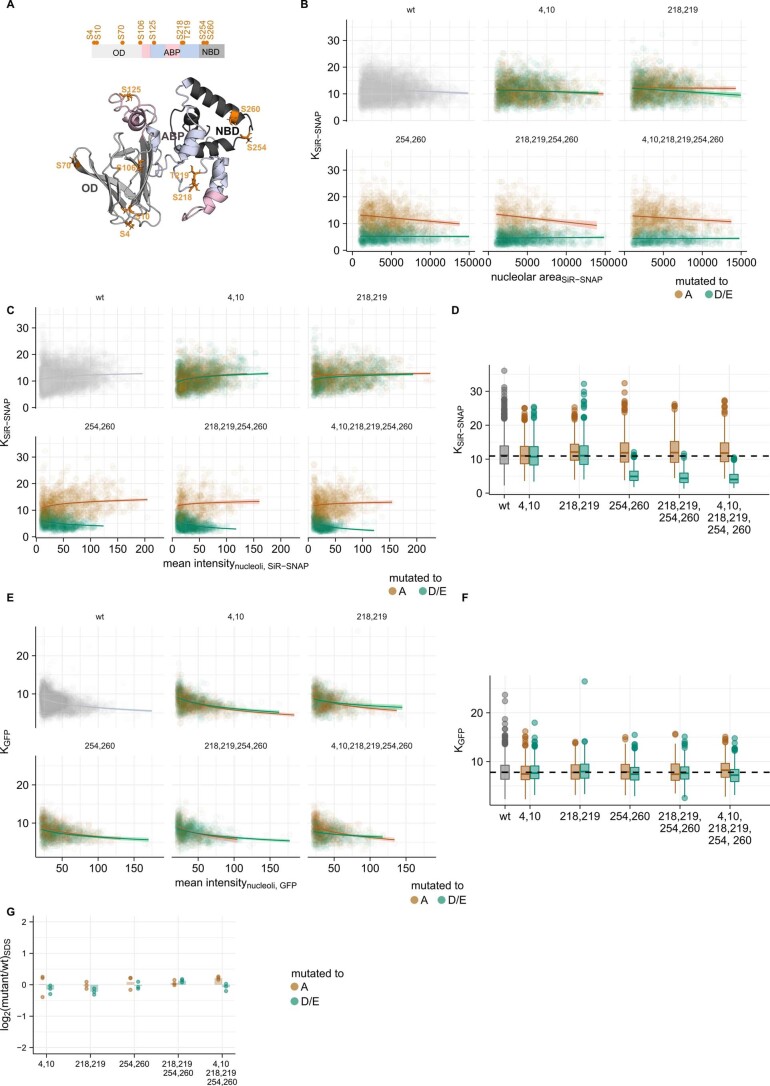
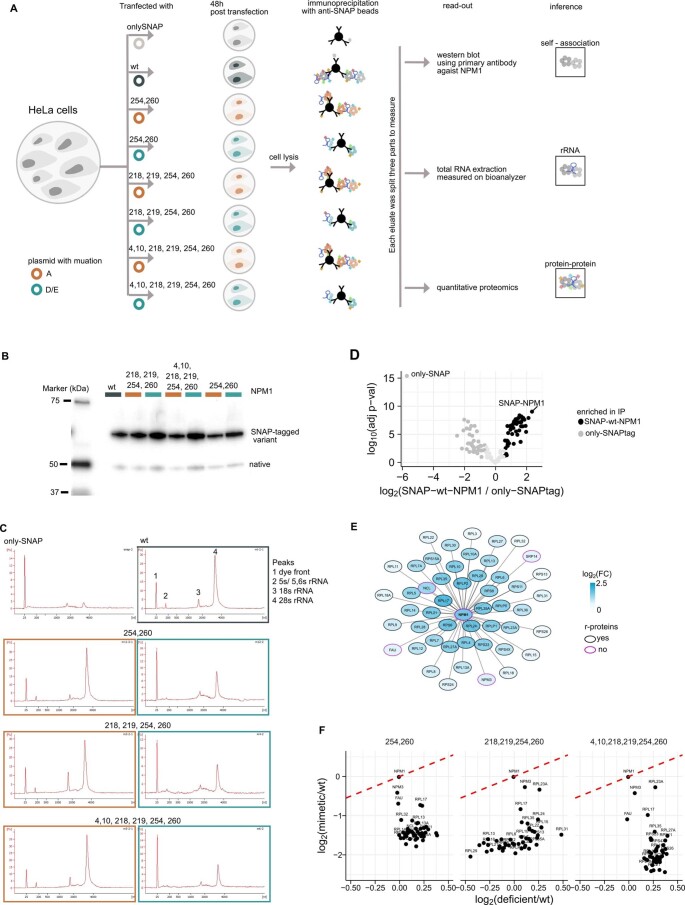
Reversible protein phosphorylation is an important mechanism for regulating (dis)assembly of biomolecular condensates. However, condensate-specific phosphosites remain largely unknown, thereby limiting our understanding of the underlying mechanisms. Here, we combine solubility proteome profiling with phosphoproteomics to quantitatively map several hundred phosphosites enriched in either soluble or condensate-bound protein subpopulations, including a subset of phosphosites modulating protein-RNA interactions. We show that multi-phosphorylation of the C-terminal disordered segment of heteronuclear ribonucleoprotein A1 (HNRNPA1), a key RNA-splicing factor, reduces its ability to locate to nuclear clusters. For nucleophosmin 1 (NPM1), an essential nucleolar protein, we show that phosphorylation of S254 and S260 is crucial for lowering its partitioning to the nucleolus and additional phosphorylation of distal sites enhances its retention in the nucleoplasm. These phosphorylation events decrease RNA and protein interactions of NPM1 to regulate its condensation. Our dataset is a rich resource for systematically uncovering the phosphoregulation of biomolecular condensates.
© 2022. The Author(s).
Conflict of interest statement
M.B. is an employer and shareholder of Cellzome, GlaxoSmithKline. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
