

Polymerogenic neuroserpin causes mitochondrial alterations and activates NFκB but not the UPR in a neuronal model of neurodegeneration FENIB
- PMID: 35864382
- PMCID: PMC9304071
- DOI: 10.1007/s00018-022-04463-3
Polymerogenic neuroserpin causes mitochondrial alterations and activates NFκB but not the UPR in a neuronal model of neurodegeneration FENIB
Abstract
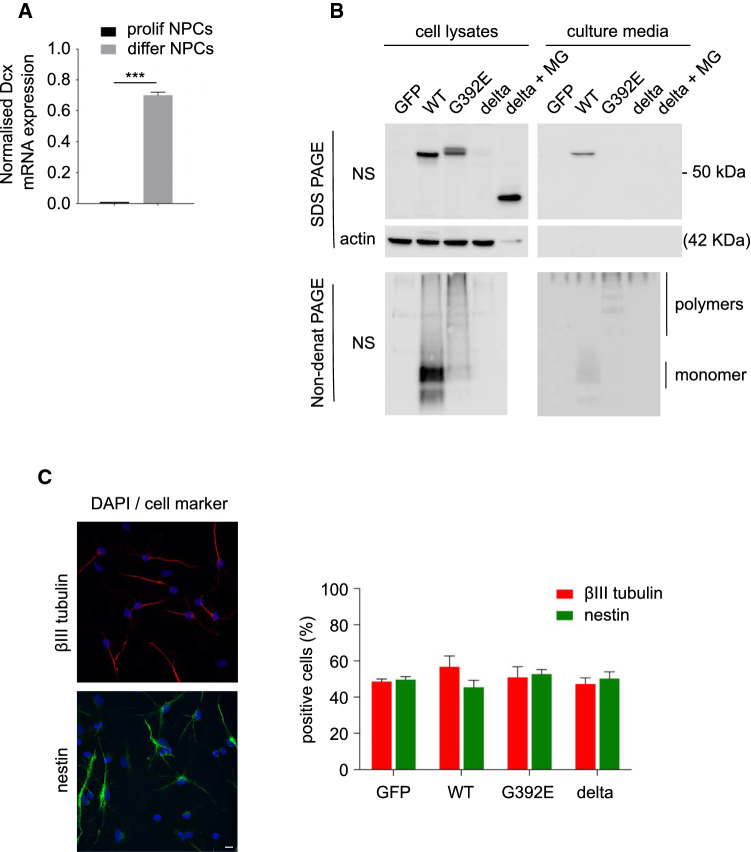
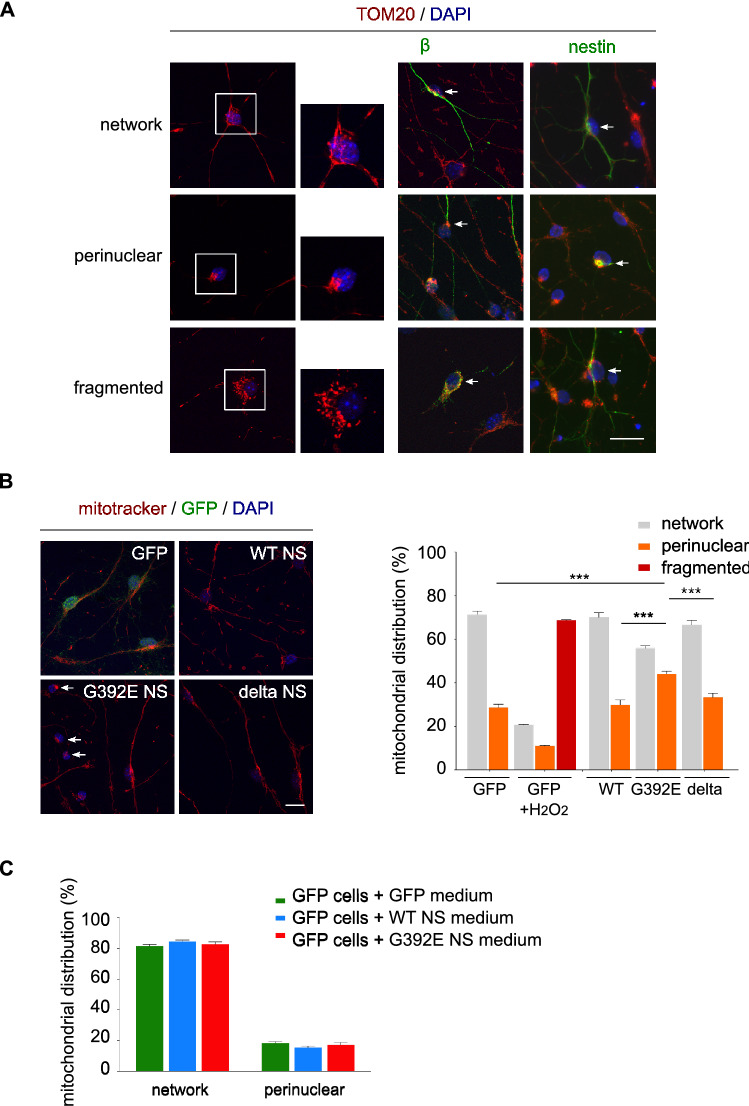
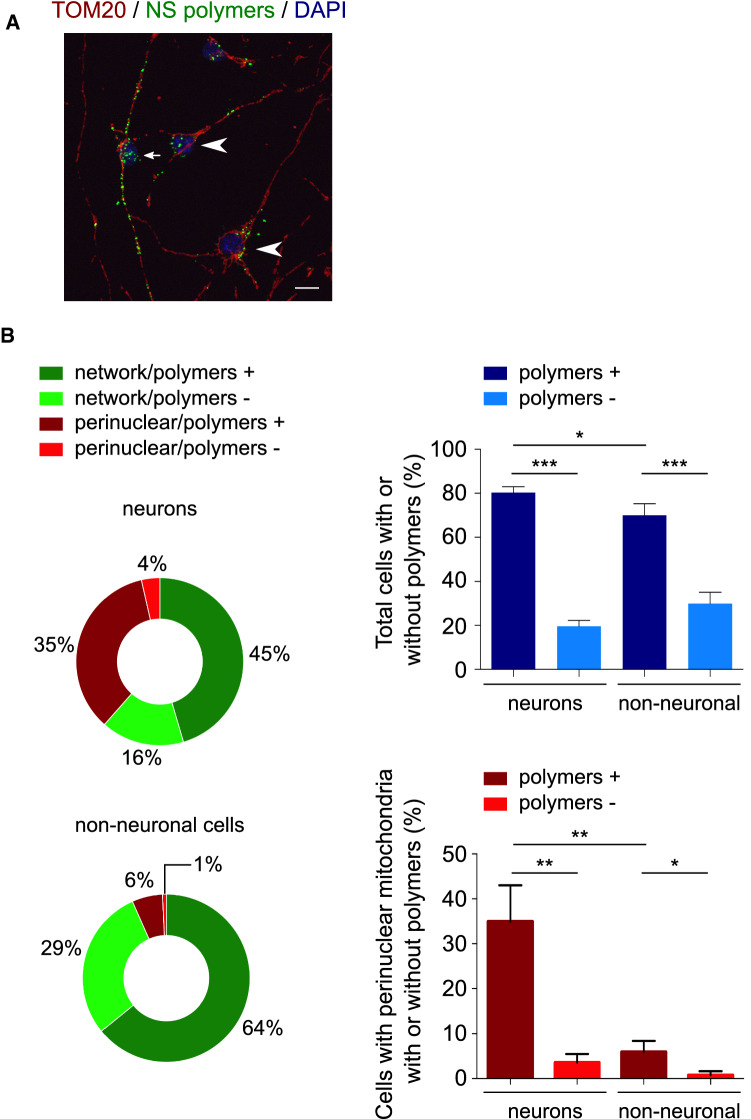
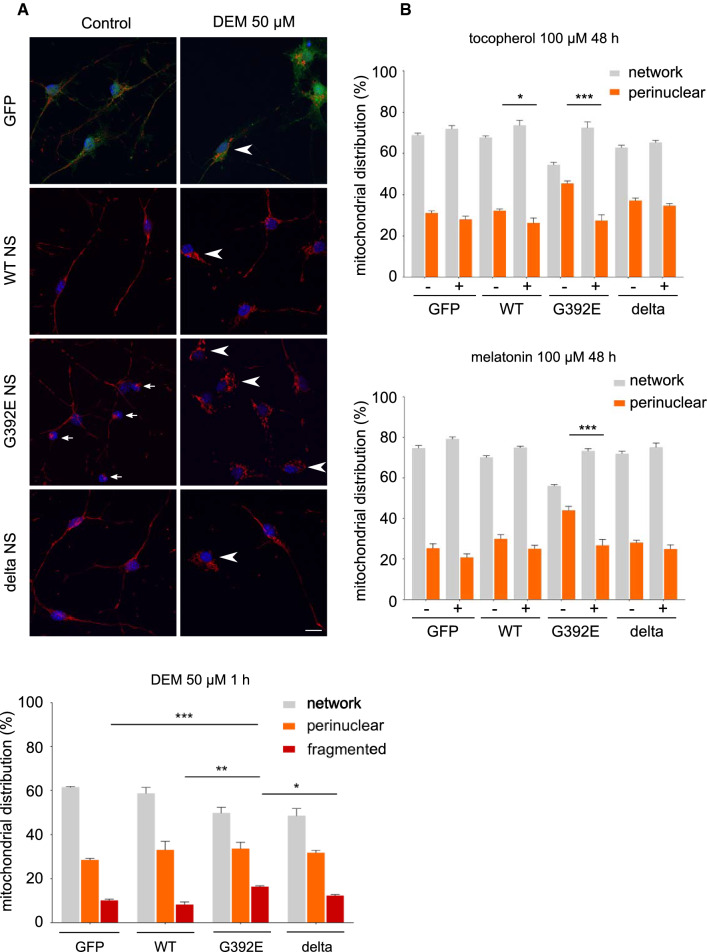
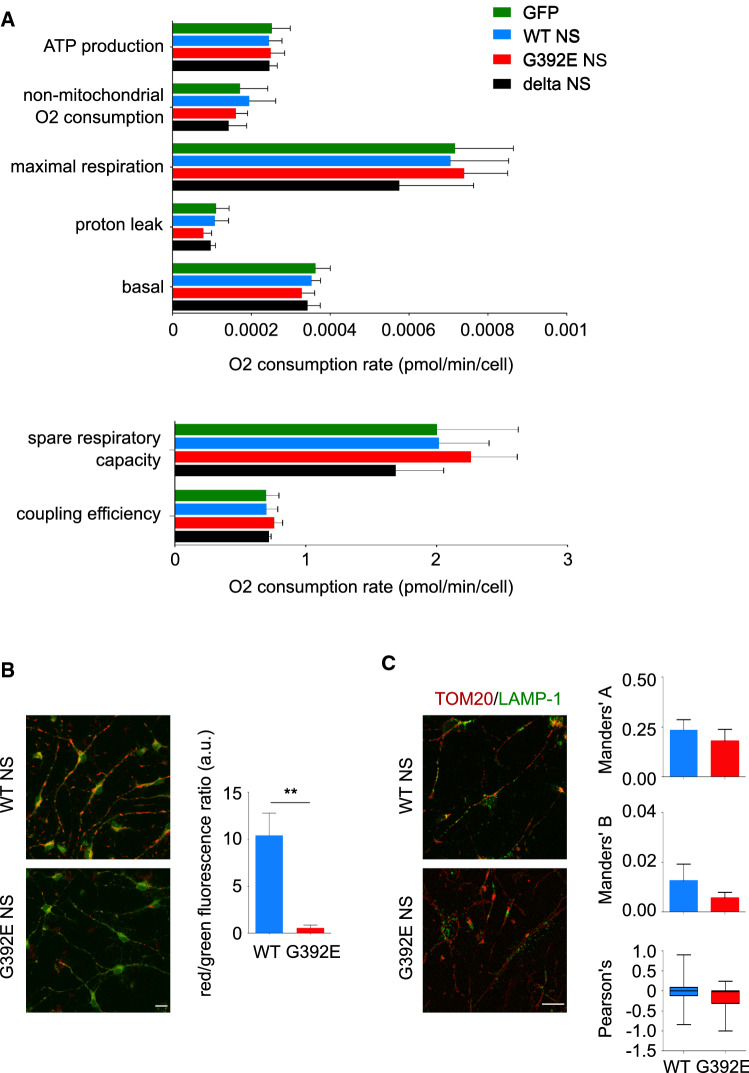
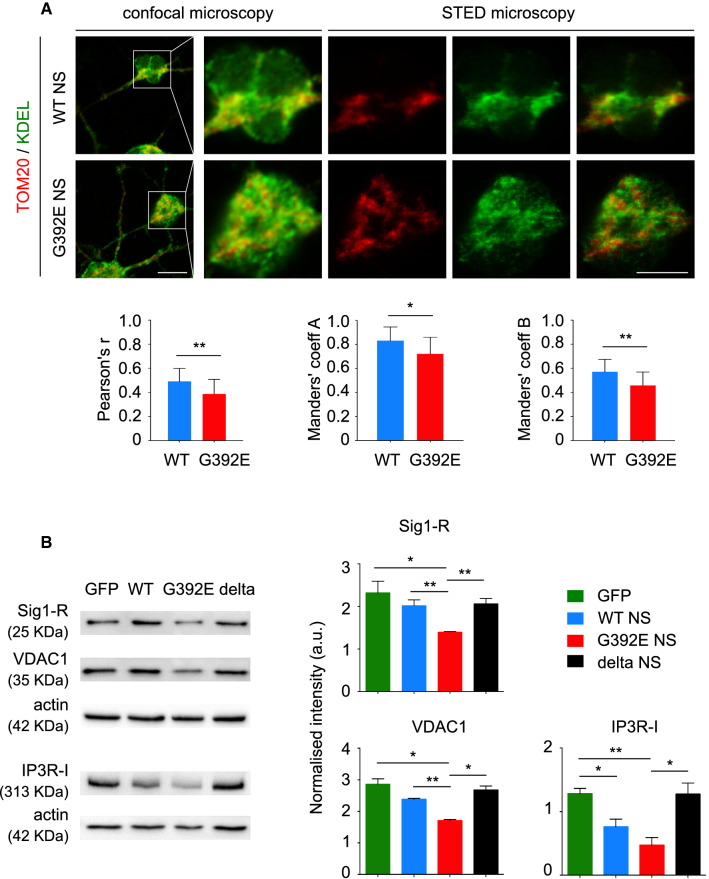
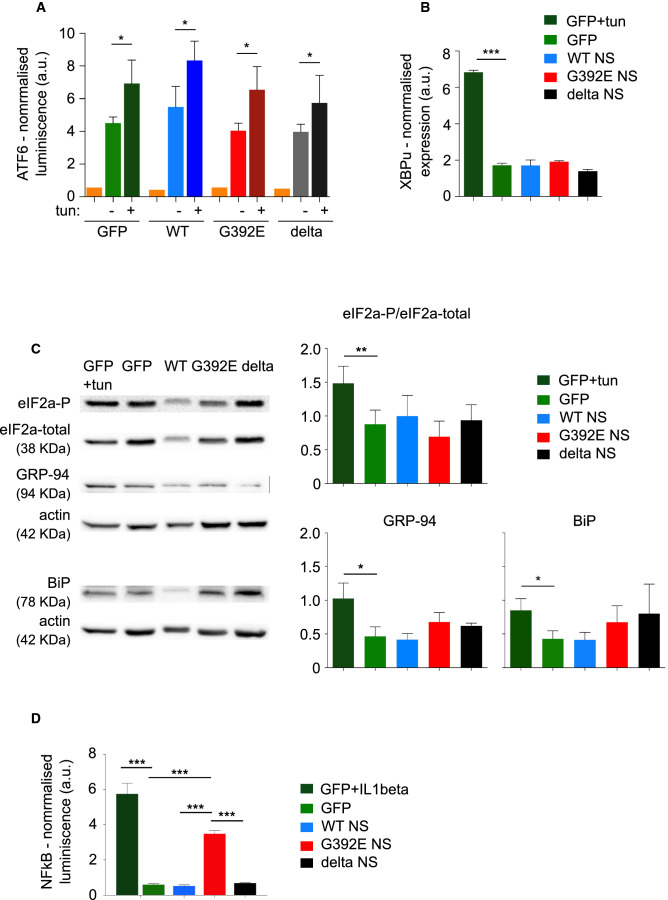
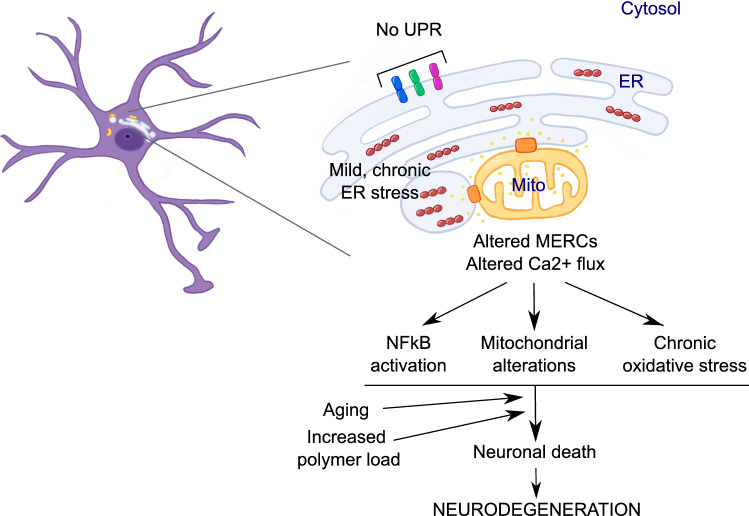
The neurodegenerative condition FENIB (familiar encephalopathy with neuroserpin inclusion bodies) is caused by heterozygous expression of polymerogenic mutant neuroserpin (NS), with polymer deposition within the endoplasmic reticulum (ER) of neurons. We generated transgenic neural progenitor cells (NPCs) from mouse fetal cerebral cortex stably expressing either the control protein GFP or human wild type, polymerogenic G392E or truncated (delta) NS. This cellular model makes it possible to study the toxicity of polymerogenic NS in the appropriated cell type by in vitro differentiation to neurons. Our previous work showed that expression of G392E NS in differentiated NPCs induced an adaptive response through the upregulation of several genes involved in the defence against oxidative stress, and that pharmacological reduction of the antioxidant defences by drug treatments rendered G392E NS neurons more susceptible to apoptosis than control neurons. In this study, we assessed mitochondrial distribution and found a higher percentage of perinuclear localisation in G392E NS neurons, particularly in those containing polymers, a phenotype that was enhanced by glutathione chelation and rescued by antioxidant molecules. Mitochondrial membrane potential and contact sites between mitochondria and the ER were reduced in neurons expressing the G392E mutation. These alterations were associated with a pattern of ER stress that involved the ER overload response but not the unfolded protein response. Our results suggest that intracellular accumulation of NS polymers affects the interaction between the ER and mitochondria, causing mitochondrial alterations that contribute to the neuronal degeneration seen in FENIB patients.
Keywords: ER overload; MERCs; Neural progenitor cells; Neurodegeneration; Neurons; Oxidative stress; Protein conformational disease; Serpin polymers.
© 2022. The Author(s).
Conflict of interest statement
The authors have no relevant financial or non-financial interests to disclose.
Figures
References
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
