

Drug Resistance Mechanisms of Acute Myeloid Leukemia Stem Cells
- PMID: 35865470
- PMCID: PMC9294245
- DOI: 10.3389/fonc.2022.896426
Drug Resistance Mechanisms of Acute Myeloid Leukemia Stem Cells
Abstract
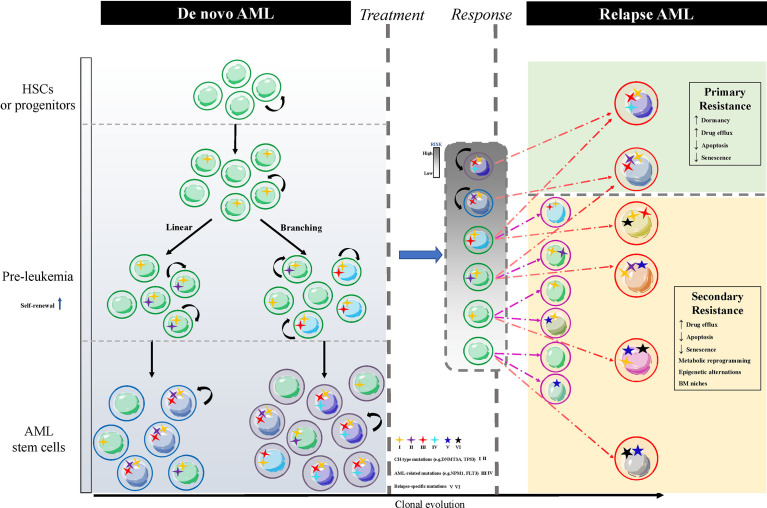
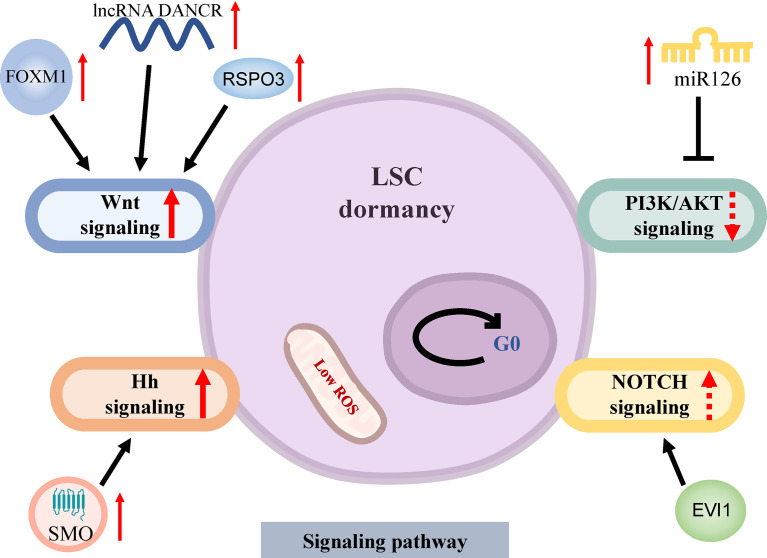
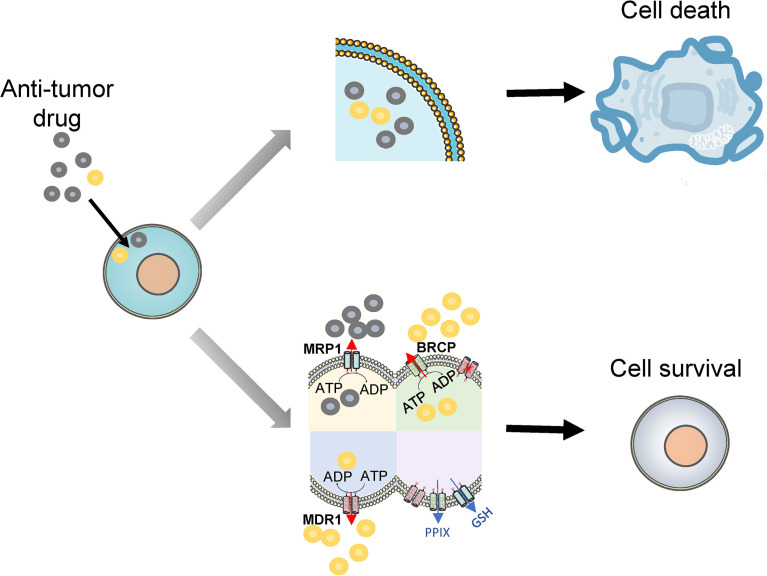
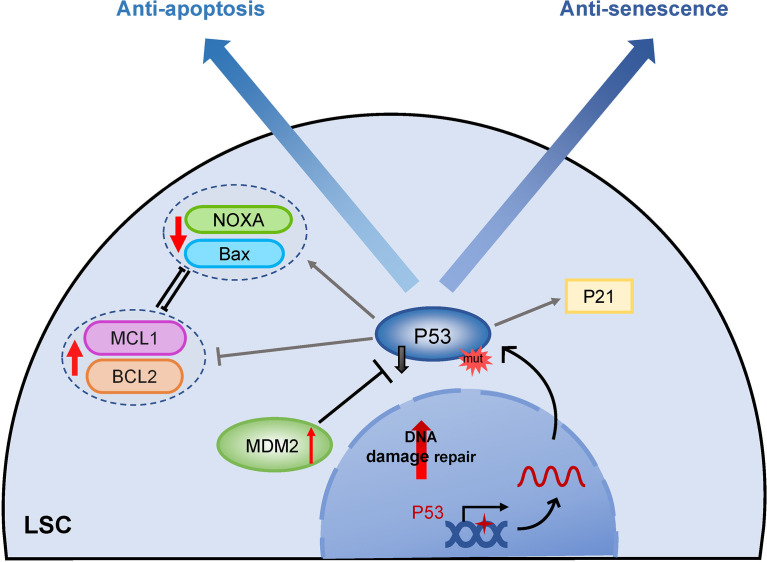
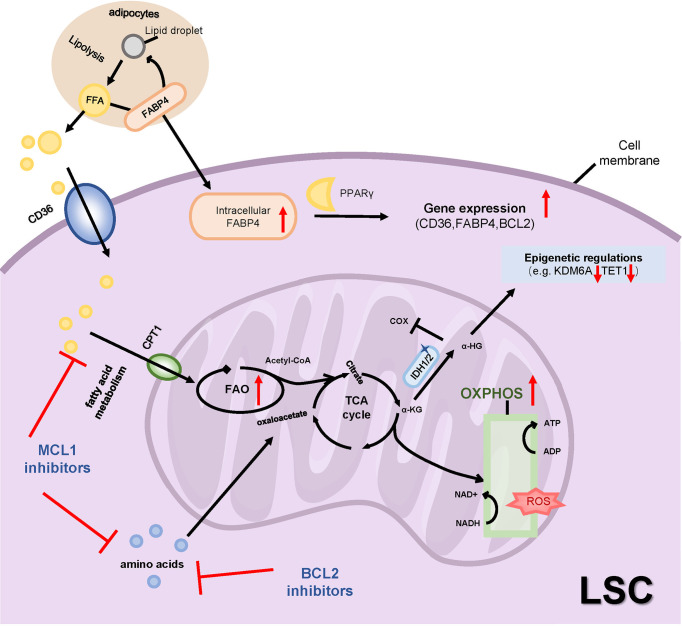
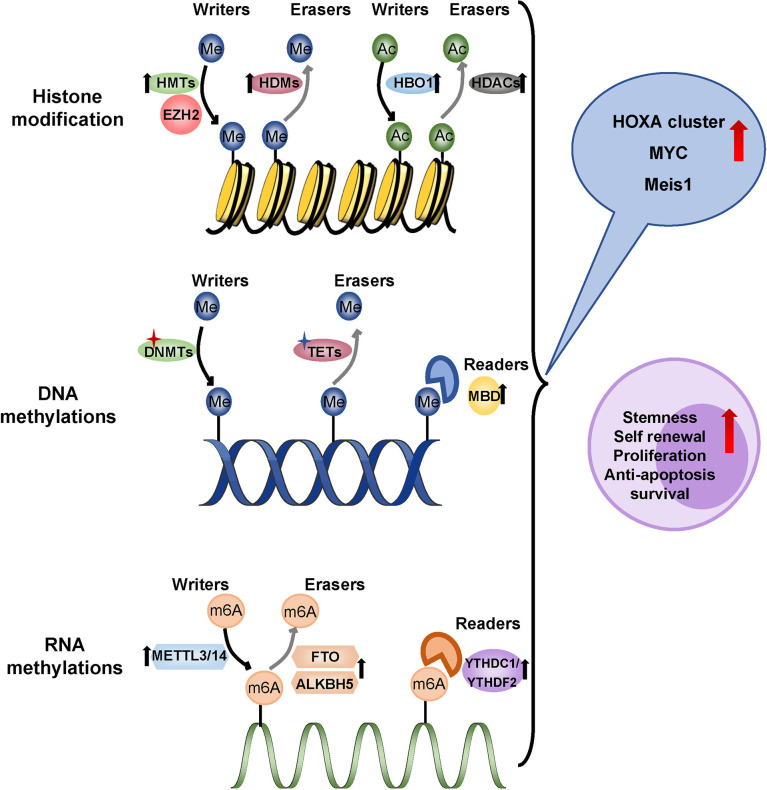
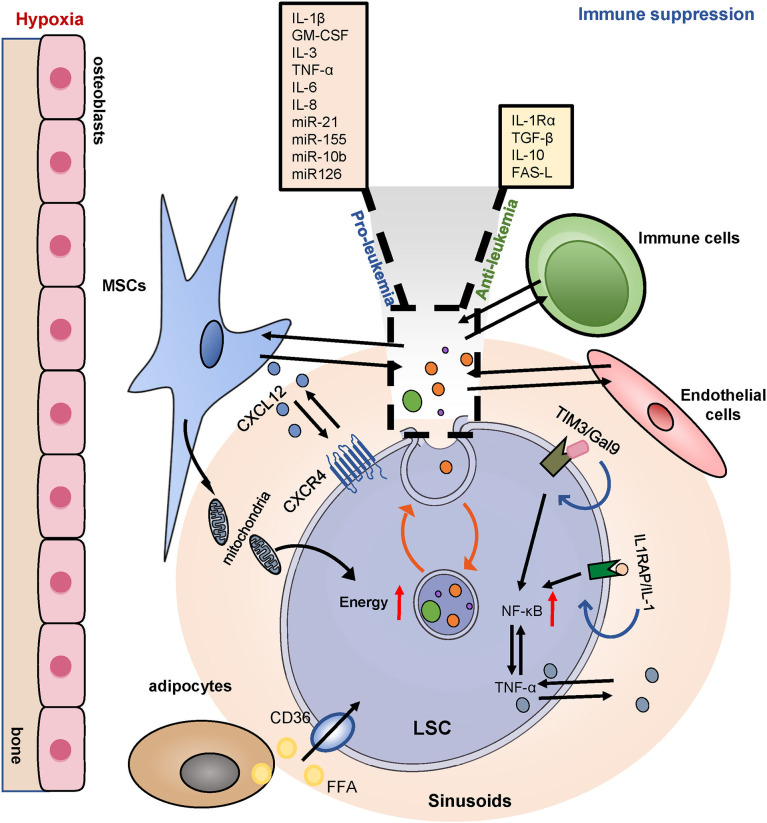
Acute myeloid leukemia (AML) is a polyclonal and heterogeneous hematological malignancy. Relapse and refractory after induction chemotherapy are still challenges for curing AML. Leukemia stem cells (LSCs), accepted to originate from hematopoietic stem/precursor cells, are the main root of leukemogenesis and drug resistance. LSCs are dynamic derivations and possess various elusive resistance mechanisms. In this review, we summarized different primary resistance and remolding mechanisms of LSCs after chemotherapy, as well as the indispensable role of the bone marrow microenvironment on LSCs resistance. Through a detailed and comprehensive review of the spectacle of LSCs resistance, it can provide better strategies for future researches on eradicating LSCs and clinical treatment of AML.
Keywords: LSCs remolding; acute myeloid leukemia; clonal heterogeneity; drug resistance; leukemia stem cells; resistant mechanisms.
Copyright © 2022 Niu, Peng and Liu.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
Leukemia stem cell-bone marrow microenvironment interplay in acute myeloid leukemia development.Exp Hematol Oncol. 2021 Jul 10;10(1):39. doi: 10.1186/s40164-021-00233-2. Exp Hematol Oncol. 2021. PMID: 34246314 Free PMC article. Review.
-
Cellular and Molecular State of Myeloid Leukemia Stem Cells.Adv Exp Med Biol. 2019;1143:41-57. doi: 10.1007/978-981-13-7342-8_2. Adv Exp Med Biol. 2019. PMID: 31338814
-
The genesis and evolution of acute myeloid leukemia stem cells in the microenvironment: From biology to therapeutic targeting.Cell Death Discov. 2022 Sep 26;8(1):397. doi: 10.1038/s41420-022-01193-0. Cell Death Discov. 2022. PMID: 36163119 Free PMC article. Review.
-
Therapeutic targeting of leukemia stem cells in acute myeloid leukemia.Front Oncol. 2023 Aug 3;13:1204895. doi: 10.3389/fonc.2023.1204895. eCollection 2023. Front Oncol. 2023. PMID: 37601659 Free PMC article. Review.
-
Escape From Treatment; the Different Faces of Leukemic Stem Cells and Therapy Resistance in Acute Myeloid Leukemia.Front Oncol. 2021 May 3;11:659253. doi: 10.3389/fonc.2021.659253. eCollection 2021. Front Oncol. 2021. PMID: 34012921 Free PMC article. Review.
Cited by
-
Boswellia carterii oleoresin extracts induce caspase-mediated apoptosis and G1 cell cycle arrest in human leukaemia subtypes.Front Pharmacol. 2023 Dec 14;14:1282239. doi: 10.3389/fphar.2023.1282239. eCollection 2023. Front Pharmacol. 2023. PMID: 38155908 Free PMC article.
-
MicroRNA‑223 overexpression suppresses protein kinase C ε expression in human leukemia stem cell‑like KG‑1a cells.Mol Clin Oncol. 2024 May 28;21(1):48. doi: 10.3892/mco.2024.2746. eCollection 2024 Jul. Mol Clin Oncol. 2024. PMID: 38881704 Free PMC article.
-
Distinct N7-methylguanosine profiles of circular RNAs in drug-resistant acute myeloid leukemia.Sci Rep. 2023 Sep 7;13(1):14704. doi: 10.1038/s41598-023-41974-w. Sci Rep. 2023. PMID: 37679400 Free PMC article.
-
RAB27B-regulated exosomes mediate LSC maintenance via resistance to senescence and crosstalk with the microenvironment.Leukemia. 2024 Feb;38(2):266-280. doi: 10.1038/s41375-023-02097-3. Epub 2023 Nov 30. Leukemia. 2024. PMID: 38036630
-
Sulforaphane: An emergent anti-cancer stem cell agent.Front Oncol. 2023 Jan 23;13:1089115. doi: 10.3389/fonc.2023.1089115. eCollection 2023. Front Oncol. 2023. PMID: 36776295 Free PMC article. Review.
References
-
- Dinardo CD, Schuh AC, Stein EM, Montesinos P, Wei A, De Botton S, et al. . Effect of Enasidenib (ENA) Plus Azacitidine (AZA) on Complete Remission and Overall Response Versus AZA Monotherapy in Mutant-IDH2 (Midh2) Newly Diagnosed Acute Myeloid Leukemia (ND-AML). J Clin Oncol (2020) 38(15):7501–7501. doi: 10.1200/JCO.2020.38.15_suppl.7501 - DOI
Publication types
LinkOut - more resources
Full Text Sources