

Activin B promotes the initiation and progression of liver fibrosis
- PMID: 35866567
- PMCID: PMC9512478
- DOI: 10.1002/hep4.2037
Activin B promotes the initiation and progression of liver fibrosis
Abstract
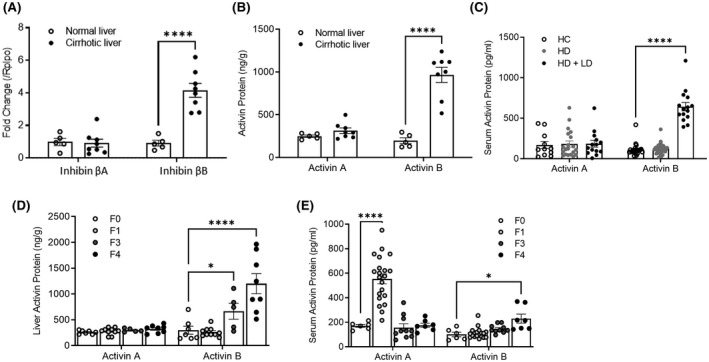
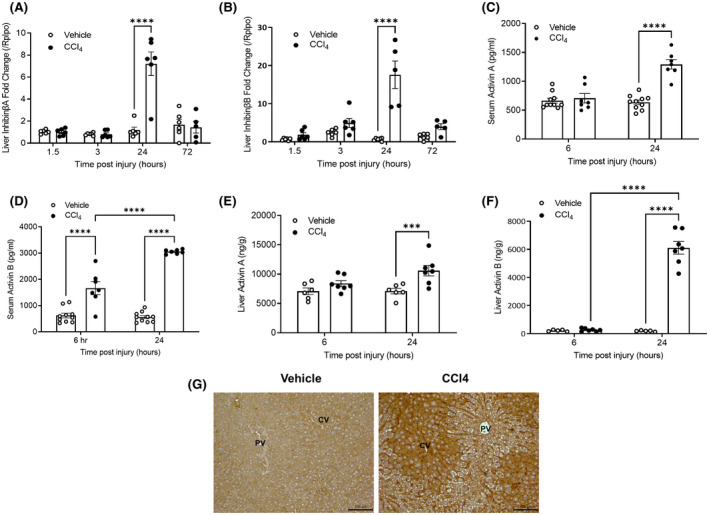
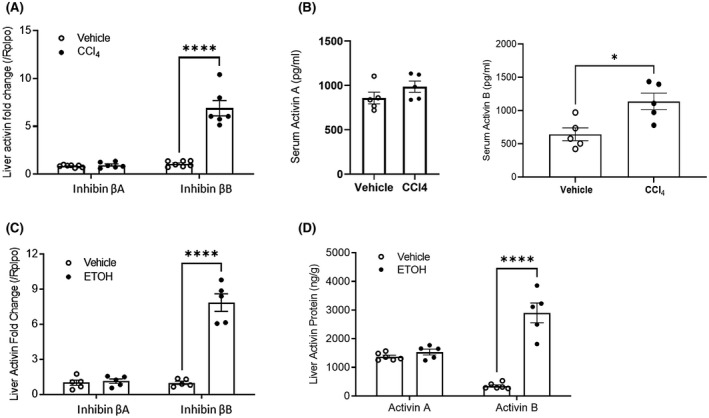
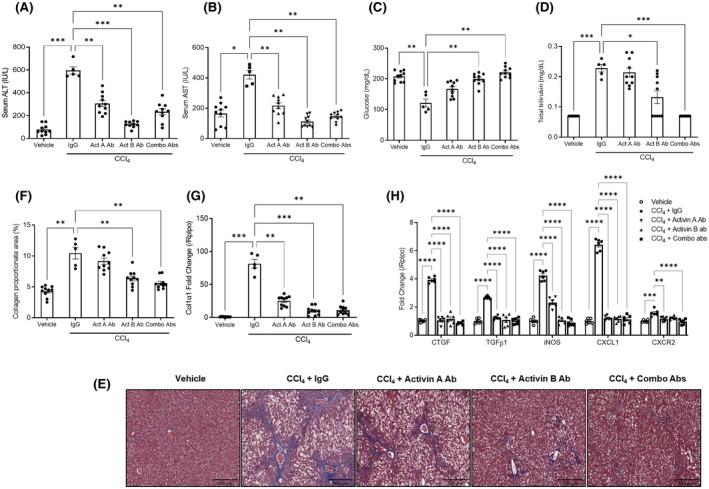
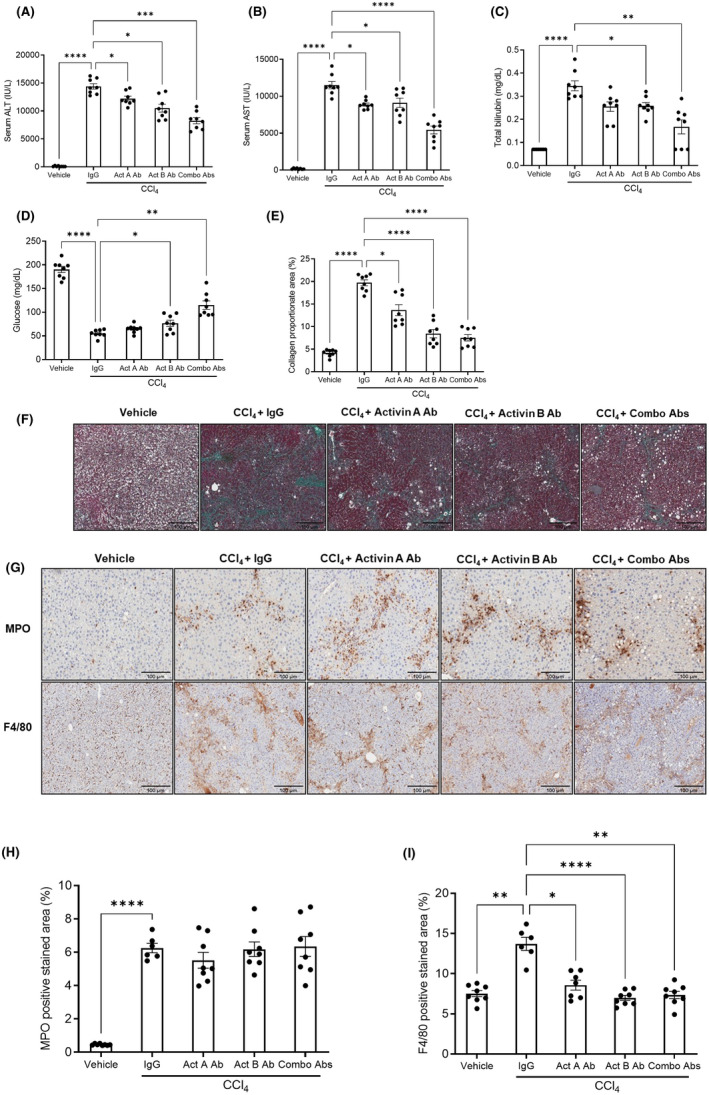
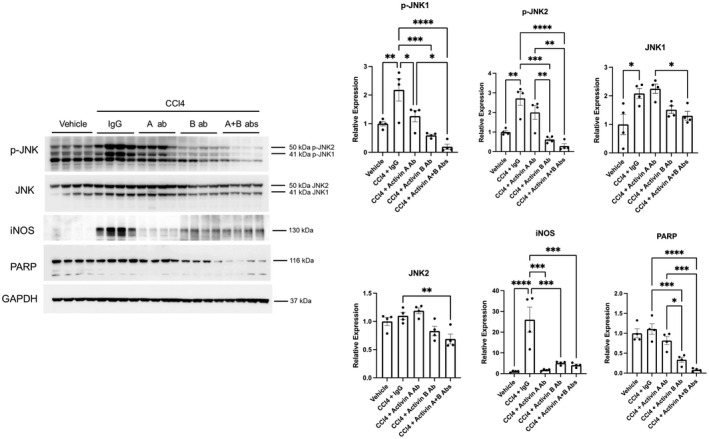
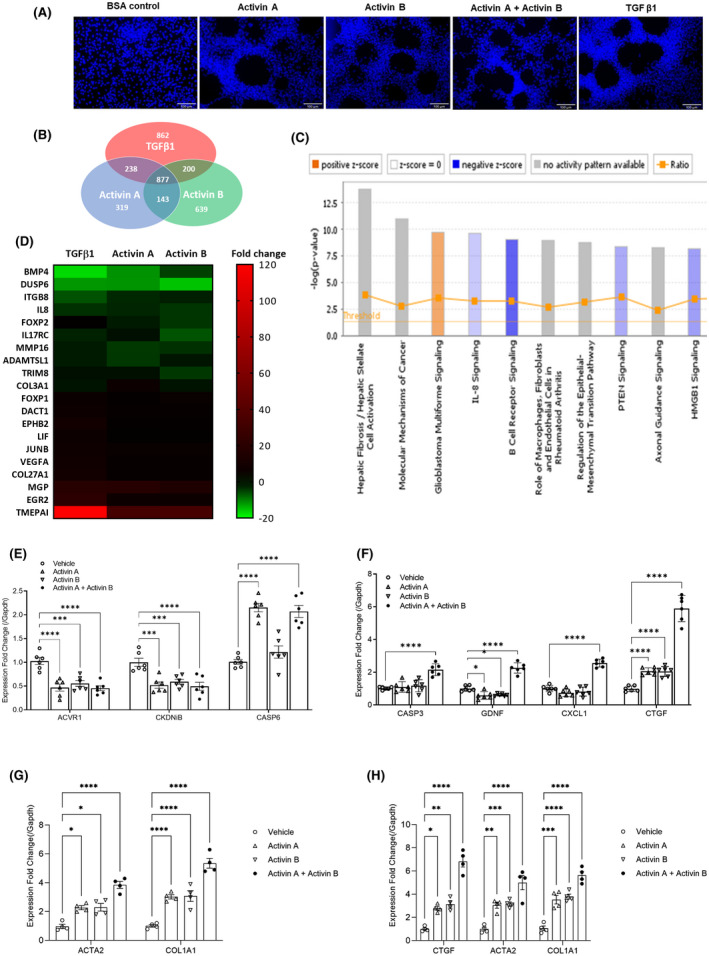
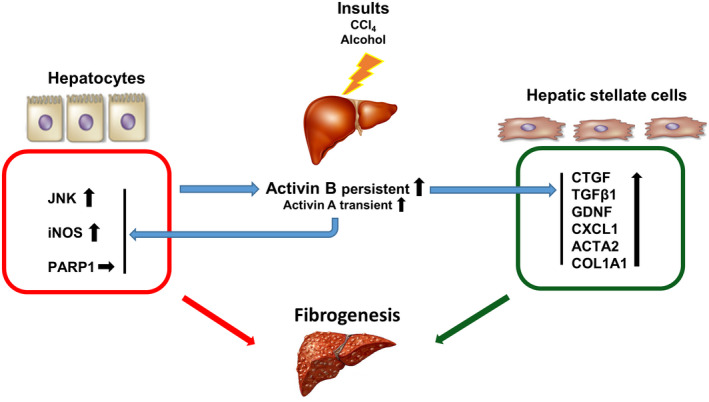
The role of activin B, a transforming growth factor β (TGFβ) superfamily cytokine, in liver health and disease is largely unknown. We aimed to investigate whether activin B modulates liver fibrogenesis. Liver and serum activin B, along with its analog activin A, were analyzed in patients with liver fibrosis from different etiologies and in mouse acute and chronic liver injury models. Activin B, activin A, or both was immunologically neutralized in mice with progressive or established carbon tetrachloride (CCl4 )-induced liver fibrosis. Hepatic and circulating activin B was increased in human patients with liver fibrosis caused by several liver diseases. In mice, hepatic and circulating activin B exhibited persistent elevation following the onset of several types of liver injury, whereas activin A displayed transient increases. The results revealed a close correlation of activin B with liver injury regardless of etiology and species. Injured hepatocytes produced excessive activin B. Neutralizing activin B largely prevented, as well as improved, CCl4 -induced liver fibrosis, which was augmented by co-neutralizing activin A. Mechanistically, activin B mediated the activation of c-Jun-N-terminal kinase (JNK), the induction of inducible nitric oxide synthase (iNOS) expression, and the maintenance of poly (ADP-ribose) polymerase 1 (PARP1) expression in injured livers. Moreover, activin B directly induced a profibrotic expression profile in hepatic stellate cells (HSCs) and stimulated these cells to form a septa structure. Conclusions: We demonstrate that activin B, cooperating with activin A, mediates the activation or expression of JNK, iNOS, and PARP1 and the activation of HSCs, driving the initiation and progression of liver fibrosis.
© 2022 The Authors. Hepatology Communications published by Wiley Periodicals LLC on behalf of American Association for the Study of Liver Diseases.
Conflict of interest statement
The authors disclose that a patent entitled “Methods of treating or preventing liver fibrosis with inhibition of activins B and A” was published based on this manuscript (patent publication number: US2021/0009672 A1). Yan Wang, Matthew Hamang, and Alexander Culver are Lilly Graduate Research Advanced Degree Program graduate students in the Department of Biology of School of Science at Indiana University–Purdue University Indianapolis and are employees of Eli Lilly & Company. Benjamin C. Yaden is an adjunct professor in the Department of Biology of School of Science at Indiana University–Purdue University Indianapolis and is an employee of Eli Lilly & Company.
Figures
Similar articles
-
Activin-A causes Hepatic stellate cell activation via the induction of TNFα and TGFβ in Kupffer cells.Biochim Biophys Acta Mol Basis Dis. 2018 Mar;1864(3):891-899. doi: 10.1016/j.bbadis.2017.12.031. Epub 2017 Dec 26. Biochim Biophys Acta Mol Basis Dis. 2018. PMID: 29287776
-
ECM1 Prevents Activation of Transforming Growth Factor β, Hepatic Stellate Cells, and Fibrogenesis in Mice.Gastroenterology. 2019 Nov;157(5):1352-1367.e13. doi: 10.1053/j.gastro.2019.07.036. Epub 2019 Jul 27. Gastroenterology. 2019. PMID: 31362006
-
Expression changes of activin A in the development of hepatic fibrosis.World J Gastroenterol. 2001 Feb;7(1):37-41. doi: 10.3748/wjg.v7.i1.37. World J Gastroenterol. 2001. PMID: 11819730 Free PMC article.
-
Thymosin β4 suppresses CCl4 -induced murine hepatic fibrosis by down-regulating transforming growth factor β receptor-II.J Gene Med. 2018 Sep;20(9):e3043. doi: 10.1002/jgm.3043. Epub 2018 Aug 15. J Gene Med. 2018. PMID: 29972714
-
A high-cholesterol diet exacerbates liver fibrosis in mice via accumulation of free cholesterol in hepatic stellate cells.Gastroenterology. 2012 Jan;142(1):152-164.e10. doi: 10.1053/j.gastro.2011.09.049. Epub 2011 Oct 10. Gastroenterology. 2012. PMID: 21995947
Cited by
-
GDF8 Contributes to Liver Fibrogenesis and Concomitant Skeletal Muscle Wasting.Biomedicines. 2023 Jul 6;11(7):1909. doi: 10.3390/biomedicines11071909. Biomedicines. 2023. PMID: 37509548 Free PMC article.
-
Single-cell multiomics guided mechanistic understanding of Fontan-associated liver disease.Sci Transl Med. 2024 Apr 24;16(744):eadk6213. doi: 10.1126/scitranslmed.adk6213. Epub 2024 Apr 24. Sci Transl Med. 2024. PMID: 38657025 Free PMC article.
-
Roles of Activin A and Gpnmb in Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD).Diabetes. 2024 Feb 1;73(2):260-279. doi: 10.2337/db23-0357. Diabetes. 2024. PMID: 37934943 Free PMC article.
-
Activin B improves glucose metabolism via induction of Fgf21 and hepatic glucagon resistance.Nat Commun. 2025 Apr 17;16(1):3678. doi: 10.1038/s41467-025-58836-w. Nat Commun. 2025. PMID: 40246973 Free PMC article.
-
Rethinking Fibrosis Regression in Alcohol-associated Liver Disease Through C/EBPβ Inhibition.Cell Mol Gastroenterol Hepatol. 2025;19(9):101541. doi: 10.1016/j.jcmgh.2025.101541. Epub 2025 Jun 6. Cell Mol Gastroenterol Hepatol. 2025. PMID: 40490018 Free PMC article. No abstract available.
References
-
- Sanyal AJ. Novel therapeutic targets for steatohepatitis. Clin Res Hepatol Gastroenterol. 2015;39(Suppl 1):S46–50. - PubMed
-
- Schuppan D, Ashfaq‐Khan M, Yang AT, Kim YO. Liver fibrosis: direct antifibrotic agents and targeted therapies. Matrix Biol. 2018;68–69:435–51. - PubMed
-
- Friedman SL. Evolving challenges in hepatic fibrosis. Nat Rev Gastroenterol Hepatol. 2010;7:425–36. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
