

Retrospective Analysis of Retroperitoneal-Abdominal-Pelvic Ganglioneuromas: An International Study by the Transatlantic Australasian Retroperitoneal Sarcoma Working Group (TARPSWG)
- PMID: 35866666
- PMCID: PMC10191524
- DOI: 10.1097/SLA.0000000000005625
Retrospective Analysis of Retroperitoneal-Abdominal-Pelvic Ganglioneuromas: An International Study by the Transatlantic Australasian Retroperitoneal Sarcoma Working Group (TARPSWG)
Abstract
Objective: The Transatlantic Australasian Retroperitoneal Sarcoma Working Group conducted a retrospective study on the disease course and clinical management of ganglioneuromas.
Background: Ganglioneuromas are rare tumors derived from neural crest cells. Data on these tumors remain limited to case reports and single-institution case series.
Methods: Patients of all ages with pathologically confirmed primary retroperitoneal, intra-abdominal, and pelvic ganglioneuromas between January 1, 2000, and January 1, 2020, were included. We examined demographic, clinicopathologic, and radiologic characteristics, as well as clinical management.
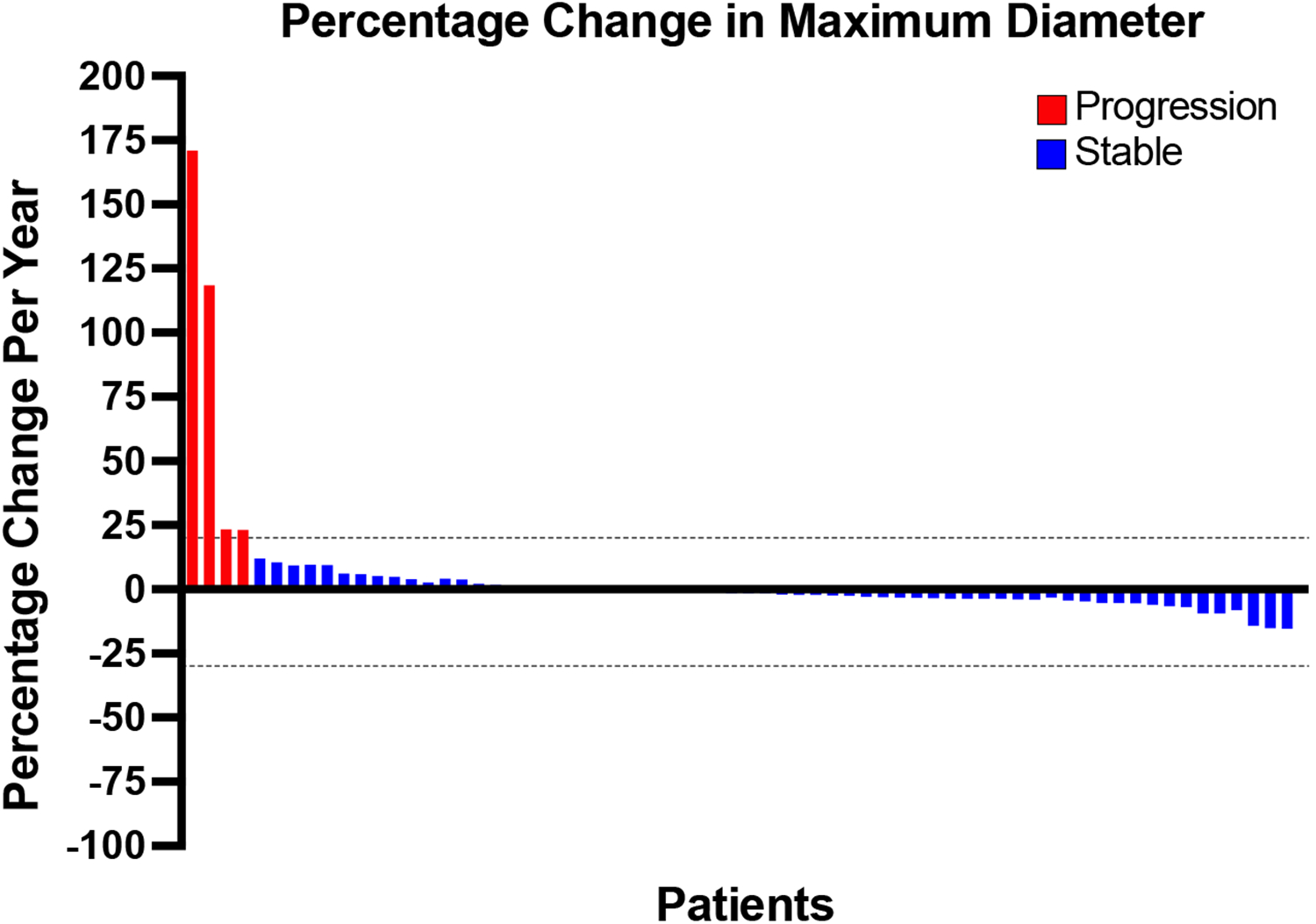
Results: Overall, 328 patients from 29 institutions were included. The median age at diagnosis was 37 years with 59.1% of patients being female. Symptomatic presentation comprised 40.9% of cases, and tumors were often located in the extra-adrenal retroperitoneum (67.1%). At baseline, the median maximum tumor diameter was 7.2 cm. One hundred sixteen (35.4%) patients underwent active surveillance, whereas 212 (64.6%) patients underwent resection with 74.5% of operative cases achieving an R0/R1 resection. Serial tumor evaluations showed that malignant transformation to neuroblastoma was rare (0.9%, N=3). Tumors undergoing surveillance had a median follow-up of 1.9 years, with 92.2% of ganglioneuromas stable in size. With a median follow-up of 3.0 years for resected tumors, 84.4% of patients were disease free after resections, whereas recurrences were observed in 4 (1.9%) patients.
Conclusions: Most ganglioneuromas have indolent disease courses and rarely transform to neuroblastoma. Thus, active surveillance may be appropriate for benign and asymptomatic tumors particularly when the risks of surgery outweigh the benefits. For symptomatic or growing tumors, resection may be curative.
Copyright © 2022 Wolters Kluwer Health, Inc. All rights reserved.
Conflict of interest statement
Conflict of Interest
The authors declare no competing interests relevant to this study.
Figures
References
-
- Geoerger B, Hero B, Harms D, et al. Metabolic activity and clinical features of primary ganglioneuromas. Cancer 2001; 91(10):1905–13. - PubMed
-
- Ambros IM, Zellner A, Roald B, et al. Role of ploidy, chromosome 1p, and Schwann cells in the maturation of neuroblastoma. N Engl J Med 1996; 334(23):1505–11. - PubMed
-
- Diab DL, Faiman C, Siperstein AE, et al. Virilizing adrenal ganglioneuroma in a woman with subclinical Cushing syndrome. Endocr Pract 2008; 14(5):584–7. - PubMed
-
- Erem C, Fidan M, Civan N, et al. Hormone-secreting large adrenal ganglioneuroma in an adult patient: a case report and review of literature. Blood Press 2014; 23(1):64–9. - PubMed
-
- Koch CA, Brouwers FM, Rosenblatt K, et al. Adrenal ganglioneuroma in a patient presenting with severe hypertension and diarrhea. Endocr Relat Cancer 2003; 10(1):99–107. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
