

Early Queen Development in Honey Bees: Social Context and Queen Breeder Source Affect Gut Microbiota and Associated Metabolism
- PMID: 35867384
- PMCID: PMC9430896
- DOI: 10.1128/spectrum.00383-22
Early Queen Development in Honey Bees: Social Context and Queen Breeder Source Affect Gut Microbiota and Associated Metabolism
Abstract
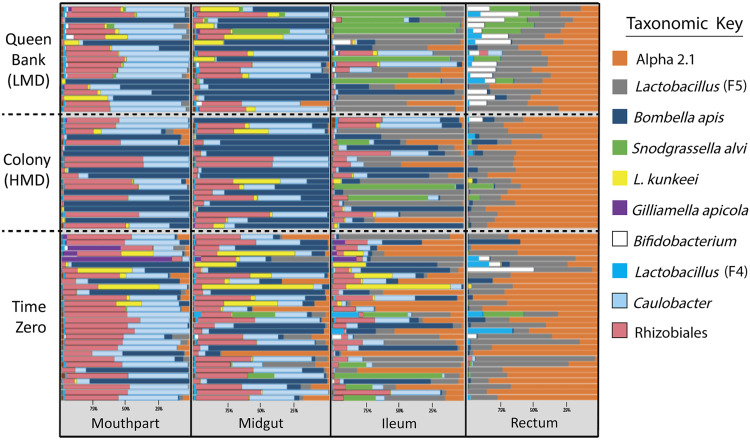
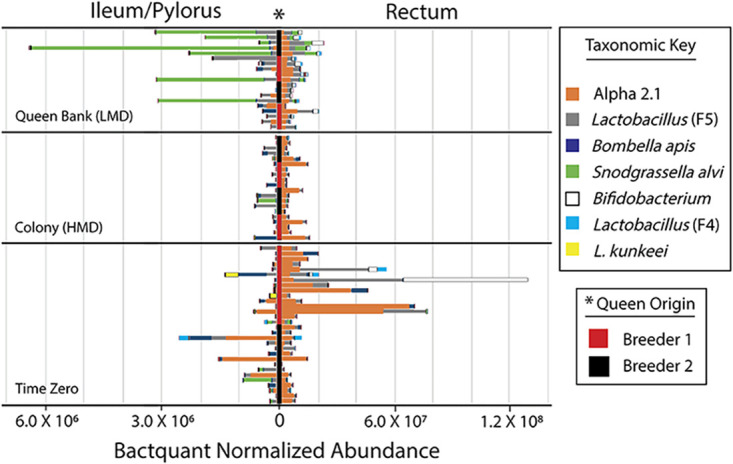
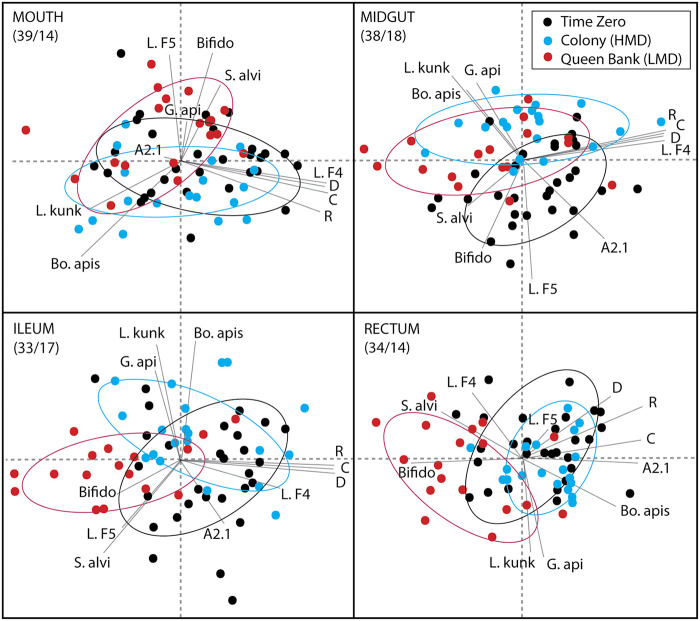
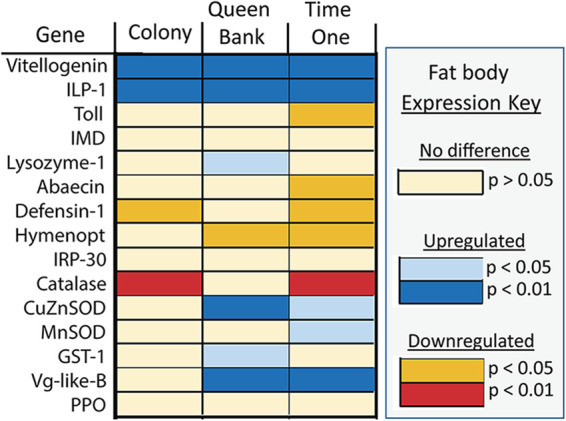
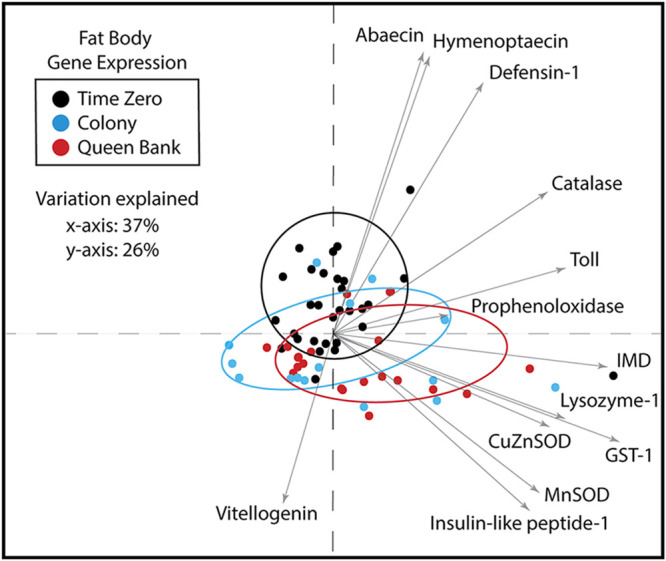
The highly social honey bee has dense populations but a significantly reduced repertoire of immune genes relative to solitary species, suggesting a greater reliance on social immunity. Here we investigate immune gene expression and gut microbial succession in queens during colony introduction. Recently mated queens were placed into an active colony or a storage hive for multiple queens: a queen-bank. Feeding intensity, social context, and metabolic demand differ greatly between the two environments. After 3 weeks, we examined gene expression associated with oxidative stress and immunity and performed high-throughput sequencing of the queen gut microbiome across four alimentary tract niches. Microbiota and gene expression in the queen hindgut differed by time, queen breeder source, and metabolic environment. In the ileum, upregulation of most immune and oxidative stress genes occurred regardless of treatment conditions, suggesting postmating effects on gut gene expression. Counterintuitively, queens exposed to the more social colony environment contained significantly less bacterial diversity indicative of social immune factors shaping the queens microbiome. Queen bank queens resembled much older queens with decreased Alpha 2.1, greater abundance of Lactobacillus firm5 and Bifidobacterium in the hindgut, and significantly larger ileum microbiotas, dominated by blooms of Snodgrassella alvi. Combined with earlier findings, we conclude that the queen gut microbiota experiences an extended period of microbial succession associated with queen breeder source, postmating development, and colony assimilation. IMPORTANCE In modern agriculture, honey bee queen failure is repeatedly cited as one of the major reasons for yearly colony loss. Here we discovered that the honey bee queen gut microbiota alters according to early social environment and is strongly tied to the identity of the queen breeder. Like human examples, this early life variation appears to set the trajectory for ecological succession associated with social assimilation and queen productivity. The high metabolic demand of natural colony assimilation is associated with less bacterial diversity, a smaller hindgut microbiome, and a downregulation of genes that control pathogens and oxidative stress. Queens placed in less social environments with low metabolic demand (queen banks) developed a gut microbiota that resembled much older queens that produce fewer eggs. The queens key reproductive role in the colony may rely in part on a gut microbiome shaped by social immunity and the early queen rearing environment.
Keywords: Apis mellifera; Bombella apis; honey bee; immune training; metabolism; microbiota; oxidative stress; queen breeder; vitellogenin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
The queen's gut refines with age: longevity phenotypes in a social insect model.Microbiome. 2018 Jun 18;6(1):108. doi: 10.1186/s40168-018-0489-1. Microbiome. 2018. PMID: 29914555 Free PMC article.
-
Metagenomic analysis of the honey bee queen microbiome reveals low bacterial diversity and Caudoviricetes phages.mSystems. 2024 Feb 20;9(2):e0118223. doi: 10.1128/msystems.01182-23. Epub 2024 Jan 23. mSystems. 2024. PMID: 38259099 Free PMC article.
-
Discovery of reproductive tissue-associated bacteria and the modes of microbiota acquisition in male honey bees (drones).mSphere. 2025 Jan 28;10(1):e0070524. doi: 10.1128/msphere.00705-24. Epub 2024 Dec 19. mSphere. 2025. PMID: 39699192 Free PMC article.
-
Structural diversity and functional variability of gut microbial communities associated with honey bees.Microb Pathog. 2020 Jan;138:103793. doi: 10.1016/j.micpath.2019.103793. Epub 2019 Oct 15. Microb Pathog. 2020. PMID: 31626917 Review.
-
Putative Drone Copulation Factors Regulating Honey Bee (Apis mellifera) Queen Reproduction and Health: A Review.Insects. 2019 Jan 8;10(1):8. doi: 10.3390/insects10010008. Insects. 2019. PMID: 30626022 Free PMC article. Review.
Cited by
-
Shotgun Metagenomics Reveals Minor Micro"bee"omes Diversity Defining Differences between Larvae and Pupae Brood Combs.Int J Mol Sci. 2024 Jan 6;25(2):741. doi: 10.3390/ijms25020741. Int J Mol Sci. 2024. PMID: 38255816 Free PMC article.
-
Changes in gut microbiota and metabolism associated with phenotypic plasticity in the honey bee Apis mellifera.Front Microbiol. 2022 Dec 9;13:1059001. doi: 10.3389/fmicb.2022.1059001. eCollection 2022. Front Microbiol. 2022. PMID: 36569094 Free PMC article.
-
A longitudinal field study of commercial honey bees shows that non-native probiotics do not rescue antibiotic treatment, and are generally not beneficial.Sci Rep. 2024 Jan 23;14(1):1954. doi: 10.1038/s41598-024-52118-z. Sci Rep. 2024. PMID: 38263184 Free PMC article.
-
Gut microbiota-driven regulation of queen bee ovarian metabolism.Microbiol Spectr. 2023 Sep 26;11(5):e0214523. doi: 10.1128/spectrum.02145-23. Online ahead of print. Microbiol Spectr. 2023. PMID: 37750696 Free PMC article.
-
Acetamiprid Exposure Disrupts Gut Microbiota in Adult and Larval Worker Honeybees (Apis mellifera L.).Insects. 2024 Nov 26;15(12):927. doi: 10.3390/insects15120927. Insects. 2024. PMID: 39769529 Free PMC article.
References
-
- Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, De Vos WM, Zucker JD, Raes J, Hansen T, Bork P, Wang J, Ehrlich SD, Pedersen O, Guedon E, Delorme C, Layec S, Khaci G, Van De Guchte M, Vandemeulebrouck G, Jamet A, Dervyn R, Sanchez N, MetaHIT consortium, et al. . 2013. Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546. doi:10.1038/nature12506. - DOI - PubMed
-
- Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI. 2013. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341:1241214. doi:10.1126/science.1241214. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
