

NGS4THAL, a One-Stop Molecular Diagnosis and Carrier Screening Tool for Thalassemia and Other Hemoglobinopathies by Next-Generation Sequencing
- PMID: 35868510
- PMCID: PMC12179521
- DOI: 10.1016/j.jmoldx.2022.06.006
NGS4THAL, a One-Stop Molecular Diagnosis and Carrier Screening Tool for Thalassemia and Other Hemoglobinopathies by Next-Generation Sequencing
Abstract
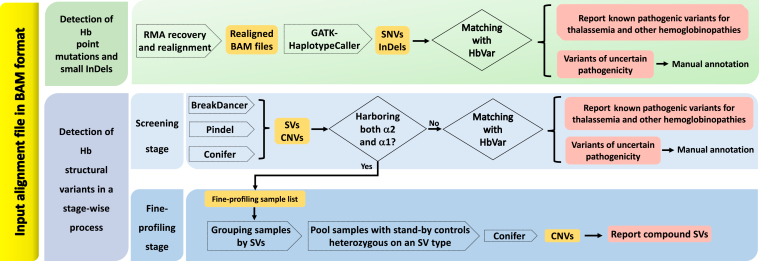
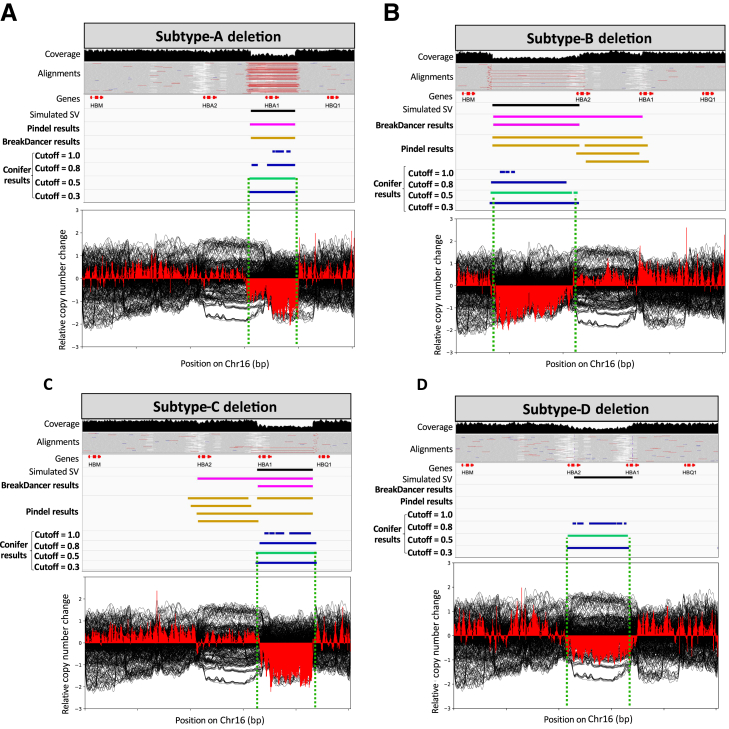
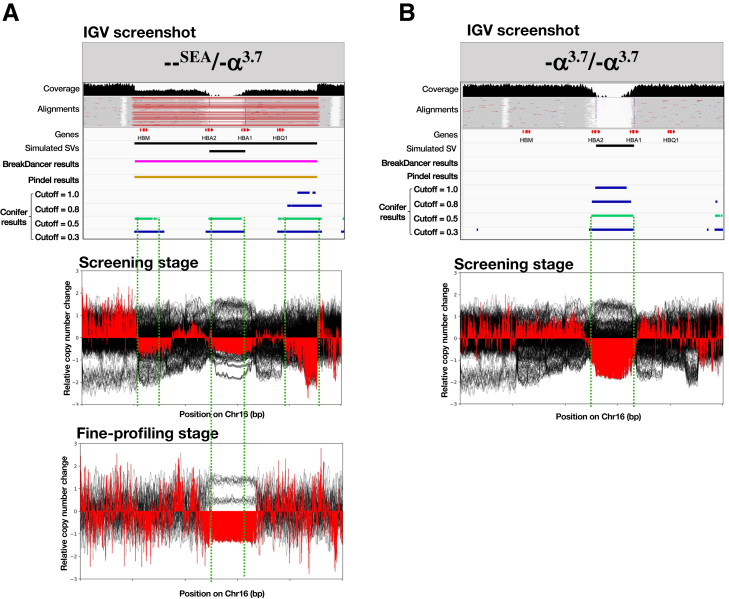
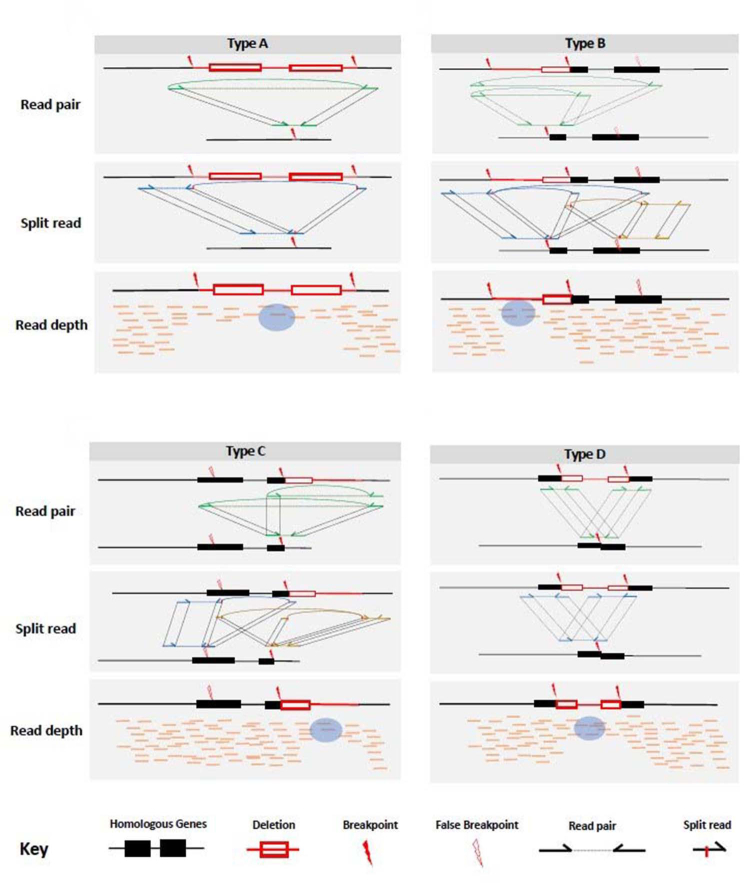
Thalassemia is one of the most common genetic diseases and a major health threat worldwide. Accurate, efficient, and scalable analysis of next-generation sequencing (NGS) data is much needed for its molecular diagnosis and carrier screening. We developed NGS4THAL, a bioinformatics analysis pipeline analyzing NGS data to detect pathogenic variants for thalassemia and other hemoglobinopathies. NGS4THAL realigns ambiguously mapped NGS reads derived from the homologous Hb gene clusters for accurate detection of point mutations and small insertions/deletions. It uses a combination of complementary structural variant (SV) detection tools and an in-house database of control data containing specific SVs to achieve accurate detection of the complex SV types. Detected variants are matched with those in HbVar (A Database of Human Hemoglobin Variants and Thalassemia Mutations), allowing recognition of known pathogenic variants, including disease modifiers. Tested on simulation data, NGS4THAL achieved high sensitivity and specificity. For targeted NGS sequencing data from samples with laboratory-confirmed pathogenic Hb variants, it achieved 100% detection accuracy. Application of NGS4THAL on whole genome sequencing data from unrelated studies revealed thalassemia mutation carrier rates for Hong Kong Chinese and Northern Vietnamese that were consistent with previous reports. NGS4THAL is a highly accurate and efficient molecular diagnosis tool for thalassemia and other hemoglobinopathies based on tailored analysis of NGS data and may be scaled for population carrier screening.
Copyright © 2022 Association for Molecular Pathology and American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures

Similar articles
-
Fetal hematological phenotypes of various hemoglobinopathies and demonstration of embryonic hemoglobins on capillary electrophoresis: a large cohort data from prenatal screening program.Diagnosis (Berl). 2025 Jan 30;12(3):441-451. doi: 10.1515/dx-2024-0190. eCollection 2025 Aug 1. Diagnosis (Berl). 2025. PMID: 39876800
-
[Expert consensus on the clinical application of Single-Molecule Real-Time Sequencing in the precise prevention and control of Thalassemia (2025 Edition)].Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025 Apr 10;42(4):385-396. doi: 10.3760/cma.j.cn511374-20250322-00173. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2025. PMID: 40555650 Chinese.
-
Detection of Common α-Hemoglobin Variants in Thailand by Using Real-Time PCR with High Resolution Melting Analysis.Hemoglobin. 2025 Jul;49(4):257-267. doi: 10.1080/03630269.2025.2528728. Epub 2025 Jul 20. Hemoglobin. 2025. PMID: 40685523
-
Pharmacological and electronic cigarette interventions for smoking cessation in adults: component network meta-analyses.Cochrane Database Syst Rev. 2023 Sep 12;9(9):CD015226. doi: 10.1002/14651858.CD015226.pub2. Cochrane Database Syst Rev. 2023. PMID: 37696529 Free PMC article.
-
Assessing the comparative effects of interventions in COPD: a tutorial on network meta-analysis for clinicians.Respir Res. 2024 Dec 21;25(1):438. doi: 10.1186/s12931-024-03056-x. Respir Res. 2024. PMID: 39709425 Free PMC article. Review.
Cited by
-
Applications of next generation sequencing in the screening and diagnosis of thalassemia: A mini-review.Front Pediatr. 2022 Sep 29;10:1015769. doi: 10.3389/fped.2022.1015769. eCollection 2022. Front Pediatr. 2022. PMID: 36245713 Free PMC article. Review.
-
Next-Generation Sequencing (NGS) and Third-Generation Sequencing (TGS) for the Diagnosis of Thalassemia.Diagnostics (Basel). 2023 Jan 19;13(3):373. doi: 10.3390/diagnostics13030373. Diagnostics (Basel). 2023. PMID: 36766477 Free PMC article. Review.
-
Comprehensive analysis of recessive carrier status using exome and genome sequencing data in 1543 Southern Chinese.NPJ Genom Med. 2022 Mar 21;7(1):23. doi: 10.1038/s41525-022-00287-z. NPJ Genom Med. 2022. PMID: 35314707 Free PMC article.
-
Variant Impact Predictor database (VIPdb), version 2: Trends from 25 years of genetic variant impact predictors.bioRxiv [Preprint]. 2024 Jun 28:2024.06.25.600283. doi: 10.1101/2024.06.25.600283. bioRxiv. 2024. Update in: Hum Genomics. 2024 Aug 28;18(1):90. doi: 10.1186/s40246-024-00663-z. PMID: 38979289 Free PMC article. Updated. Preprint.
-
Genome-wide association study of BNT162b2 vaccine-related myocarditis identifies potential predisposing functional areas in Hong Kong adolescents.BMC Genom Data. 2024 Jun 6;25(1):51. doi: 10.1186/s12863-024-01238-6. BMC Genom Data. 2024. PMID: 38844841 Free PMC article.
References
-
- Piel F.B., Weatherall D.J. The α-thalassemias. N Engl J Med. 2014;371:1908–1916. - PubMed
-
- Rund D., Rachmilewitz E. β-Thalassemia. N Engl J Med. 2005;353:1135–1146. - PubMed
-
- Giardine B., Borg J., Viennas E., Pavlidis C., Moradkhani K., Joly P., Bartsakoulia M., Riemer C., Miller W., Tzimas G., Wajcman H., Hardison R.C., Patrinos G.P. Updates of the HbVar database of human hemoglobin variants and thalassemia mutations. Nucleic Acids Res. 2014;42:D1063–D1069. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
