

Gain-of-function mutations in ALPK1 cause an NF-κB-mediated autoinflammatory disease: functional assessment, clinical phenotyping and disease course of patients with ROSAH syndrome
- PMID: 35868845
- PMCID: PMC9484401
- DOI: 10.1136/annrheumdis-2022-222629
Gain-of-function mutations in ALPK1 cause an NF-κB-mediated autoinflammatory disease: functional assessment, clinical phenotyping and disease course of patients with ROSAH syndrome
Abstract
Objectives: To test the hypothesis that ROSAH (retinal dystrophy, optic nerve oedema, splenomegaly, anhidrosis and headache) syndrome, caused by dominant mutation in ALPK1, is an autoinflammatory disease.
Methods: This cohort study systematically evaluated 27 patients with ROSAH syndrome for inflammatory features and investigated the effect of ALPK1 mutations on immune signalling. Clinical, immunologic and radiographical examinations were performed, and 10 patients were empirically initiated on anticytokine therapy and monitored. Exome sequencing was used to identify a new pathogenic variant. Cytokine profiling, transcriptomics, immunoblotting and knock-in mice were used to assess the impact of ALPK1 mutations on protein function and immune signalling.
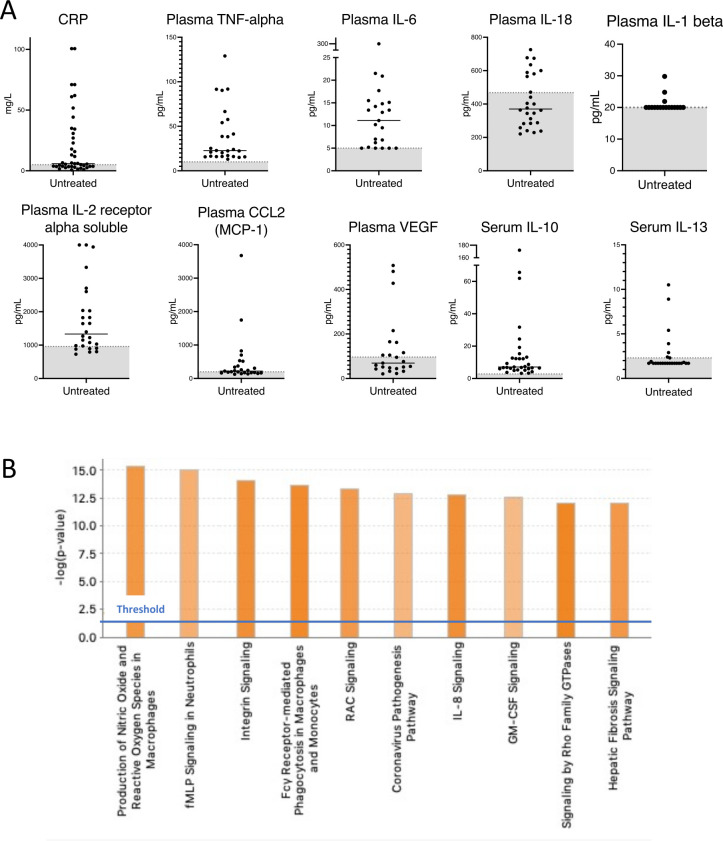
Results: The majority of the cohort carried the p.Thr237Met mutation but we also identified a new ROSAH-associated mutation, p.Tyr254Cys.Nearly all patients exhibited at least one feature consistent with inflammation including recurrent fever, headaches with meningeal enhancement and premature basal ganglia/brainstem mineralisation on MRI, deforming arthritis and AA amyloidosis. However, there was significant phenotypic variation, even within families and some adults lacked functional visual deficits. While anti-TNF and anti-IL-1 therapies suppressed systemic inflammation and improved quality of life, anti-IL-6 (tocilizumab) was the only anticytokine therapy that improved intraocular inflammation (two of two patients).Patients' primary samples and in vitro assays with mutated ALPK1 constructs showed immune activation with increased NF-κB signalling, STAT1 phosphorylation and interferon gene expression signature. Knock-in mice with the Alpk1 T237M mutation exhibited subclinical inflammation.Clinical features not conventionally attributed to inflammation were also common in the cohort and included short dental roots, enamel defects and decreased salivary flow.
Conclusion: ROSAH syndrome is an autoinflammatory disease caused by gain-of-function mutations in ALPK1 and some features of disease are amenable to immunomodulatory therapy.
Keywords: Amyloidosis; Arthritis; Immune System Diseases; Inflammation; Therapeutics.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY. Published by BMJ.
Conflict of interest statement
Competing interests: FS is a cofounder and stockholder of Pyrotech therapeutics, a company that aims to develop agonist/inhibitor drugs for ALPK1. RM has received honorary fees for lectures from SHIRE-TAKEDA- SANOFI- NOVARTIS-SOBI and Nida Sen is employed by Janssen.
Figures






References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
