

Assessing the clinical utility of protein structural analysis in genomic variant classification: experiences from a diagnostic laboratory
- PMID: 35869530
- PMCID: PMC9308257
- DOI: 10.1186/s13073-022-01082-2
Assessing the clinical utility of protein structural analysis in genomic variant classification: experiences from a diagnostic laboratory
Abstract
Background: The widespread clinical application of genome-wide sequencing has resulted in many new diagnoses for rare genetic conditions, but testing regularly identifies variants of uncertain significance (VUS). The remarkable rise in the amount of genomic data has been paralleled by a rise in the number of protein structures that are now publicly available, which may have clinical utility for the interpretation of missense and in-frame insertions or deletions.
Methods: Within a UK National Health Service genomic medicine diagnostic laboratory, we investigated the number of VUS over a 5-year period that were evaluated using protein structural analysis and how often this analysis aided variant classification.
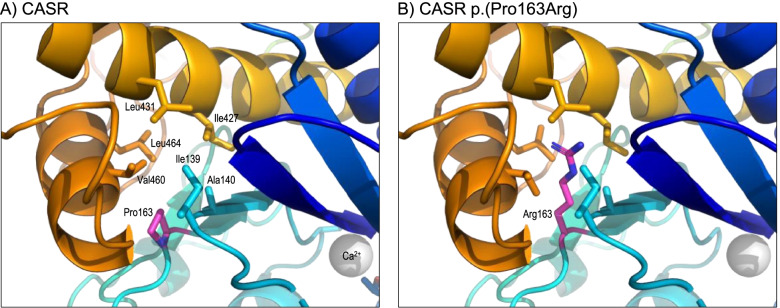
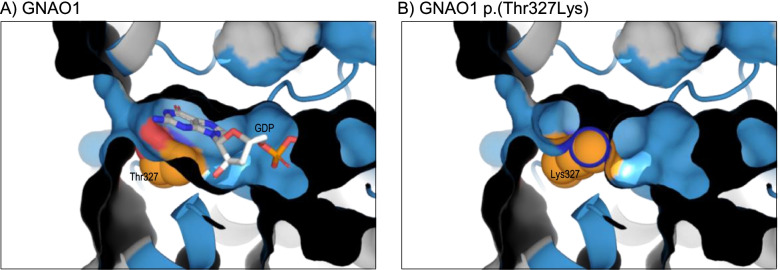
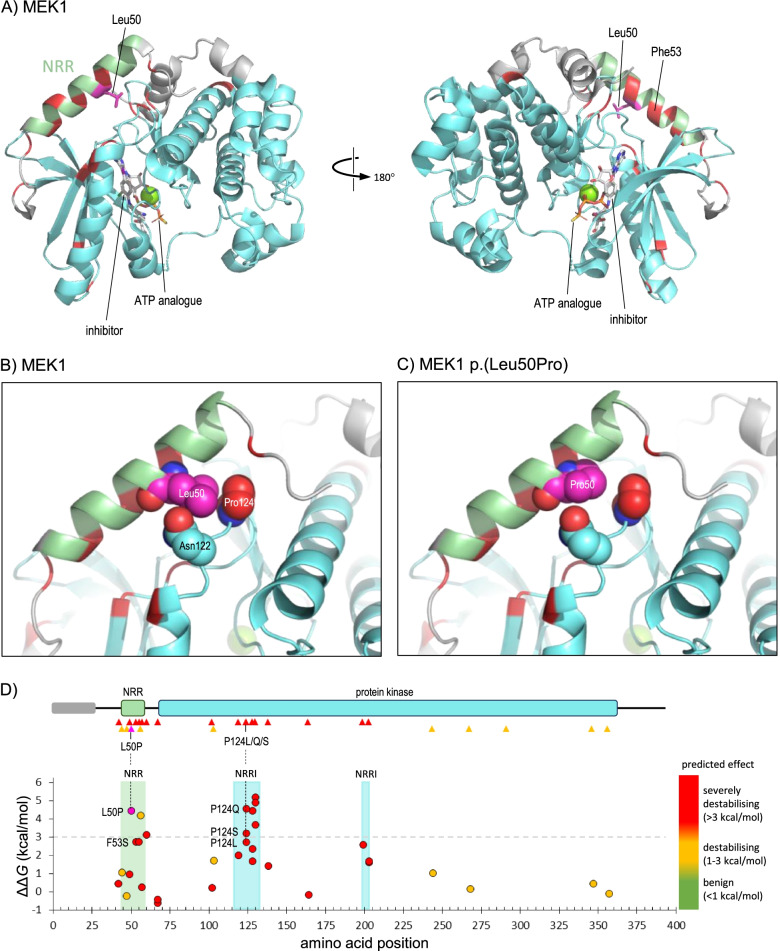
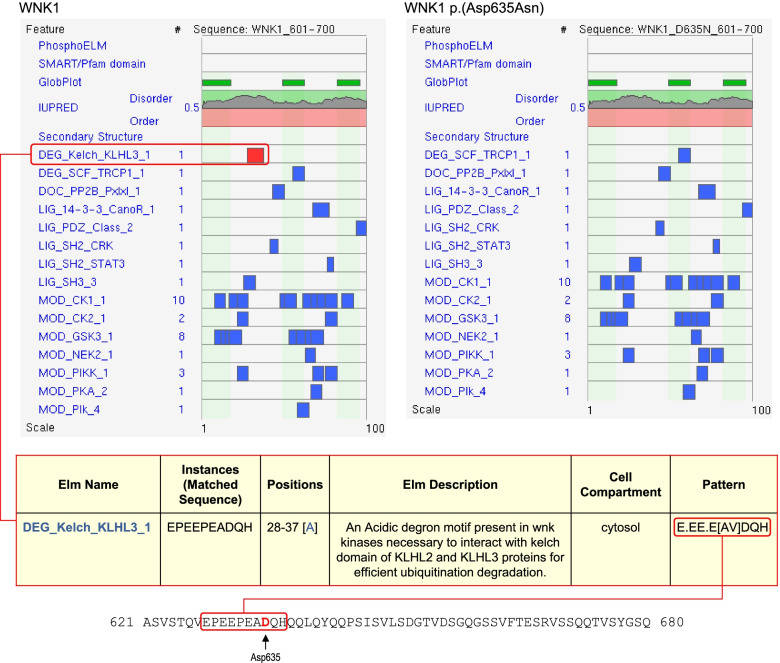
Results: We found 99 novel missense and in-frame variants across 67 genes that were initially classified as VUS by our diagnostic laboratory using standard variant classification guidelines and for which further analysis of protein structure was requested. Evidence from protein structural analysis was used in the re-assessment of 64 variants, of which 47 were subsequently reclassified as pathogenic or likely pathogenic and 17 remained as VUS. We identified several case studies where protein structural analysis aided variant interpretation by predicting disease mechanisms that were consistent with the observed phenotypes, including loss-of-function through thermodynamic destabilisation or disruption of ligand binding, and gain-of-function through de-repression or escape from proteasomal degradation.
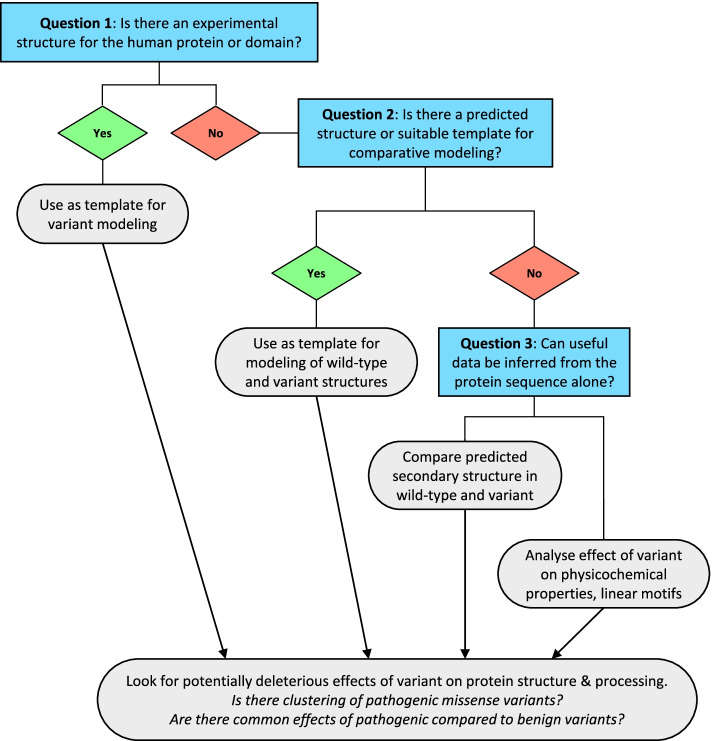
Conclusions: We have shown that using in silico protein structural analysis can aid classification of VUS and give insights into the mechanisms of pathogenicity. Based on our experience, we propose a generic evidence-based workflow for incorporating protein structural information into diagnostic practice to facilitate variant classification.
Keywords: Genomic medicine; Missense variant; Modelling; Pathogenicity; Prediction; Protein structure; Variant classification; Variant interpretation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
