

HDAC1 and PRC2 mediate combinatorial control in SPI1/PU.1-dependent gene repression in murine erythroleukaemia
- PMID: 35871293
- PMCID: PMC9371914
- DOI: 10.1093/nar/gkac613
HDAC1 and PRC2 mediate combinatorial control in SPI1/PU.1-dependent gene repression in murine erythroleukaemia
Abstract
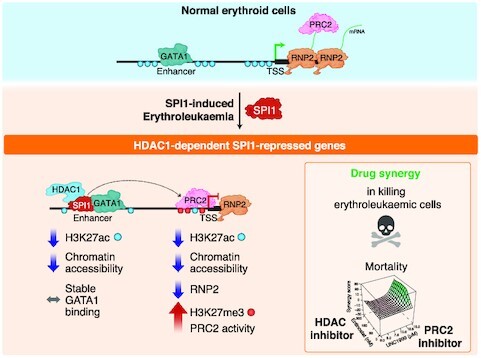
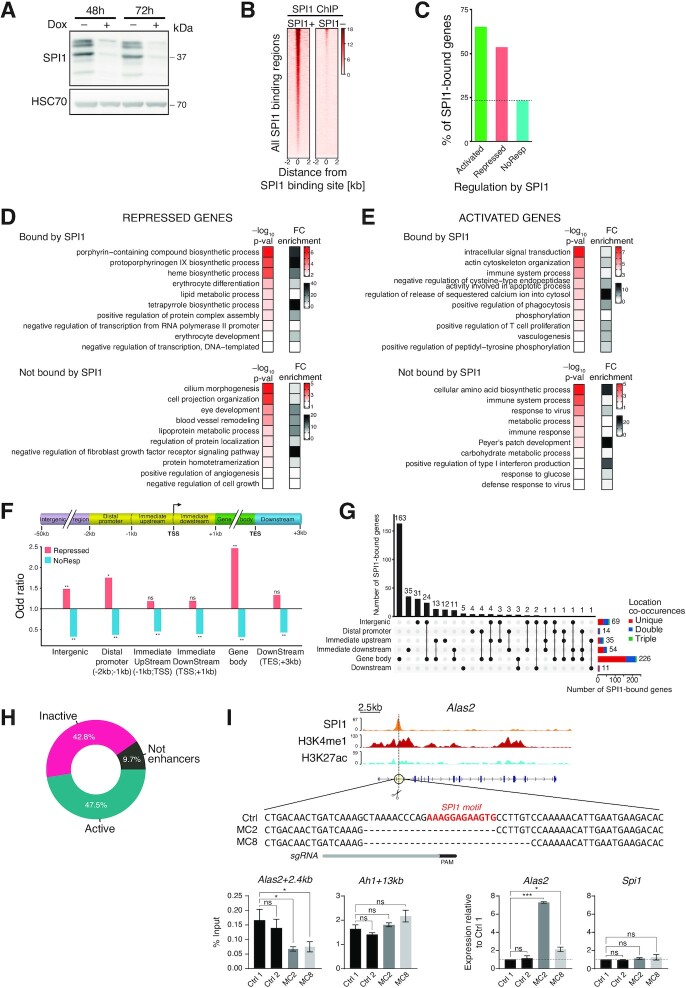
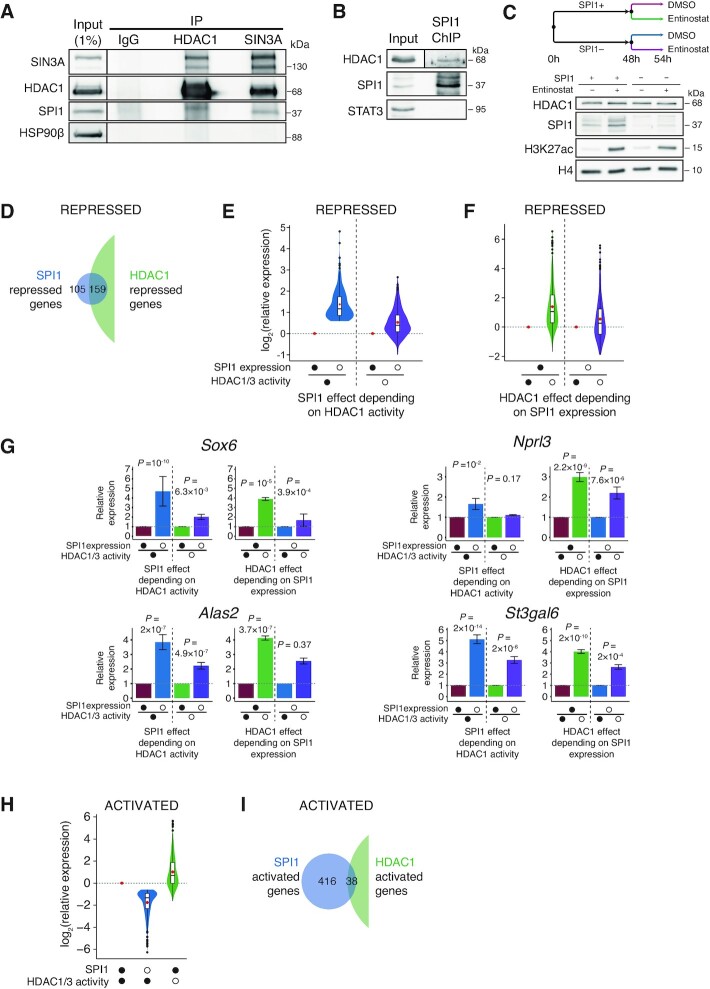
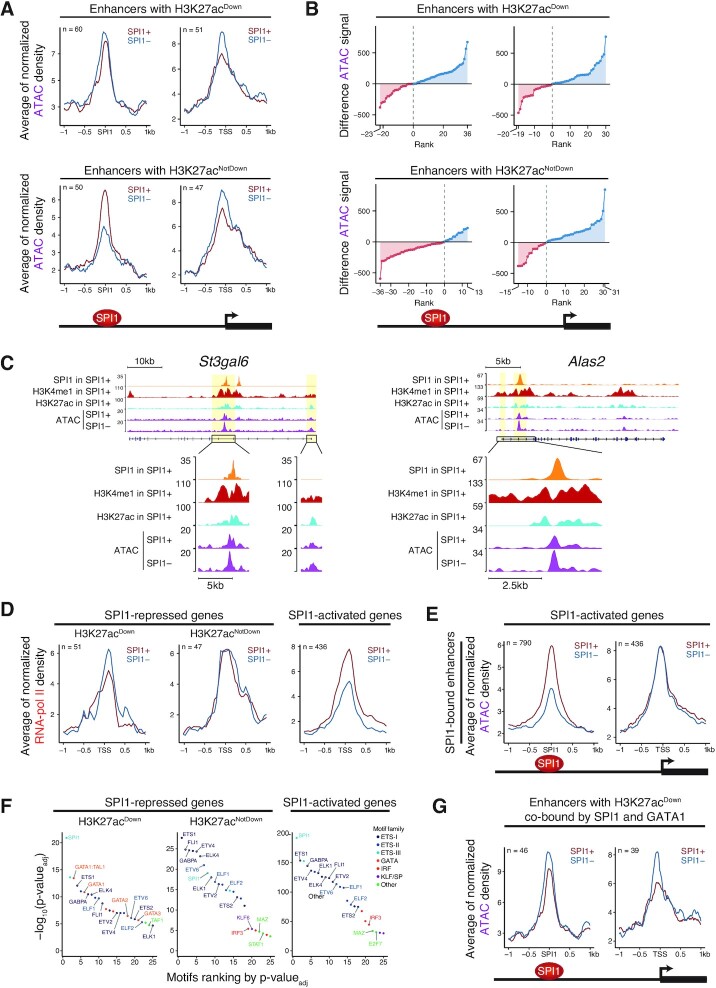
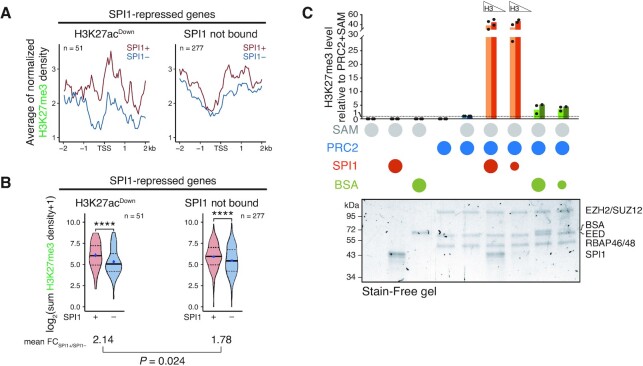
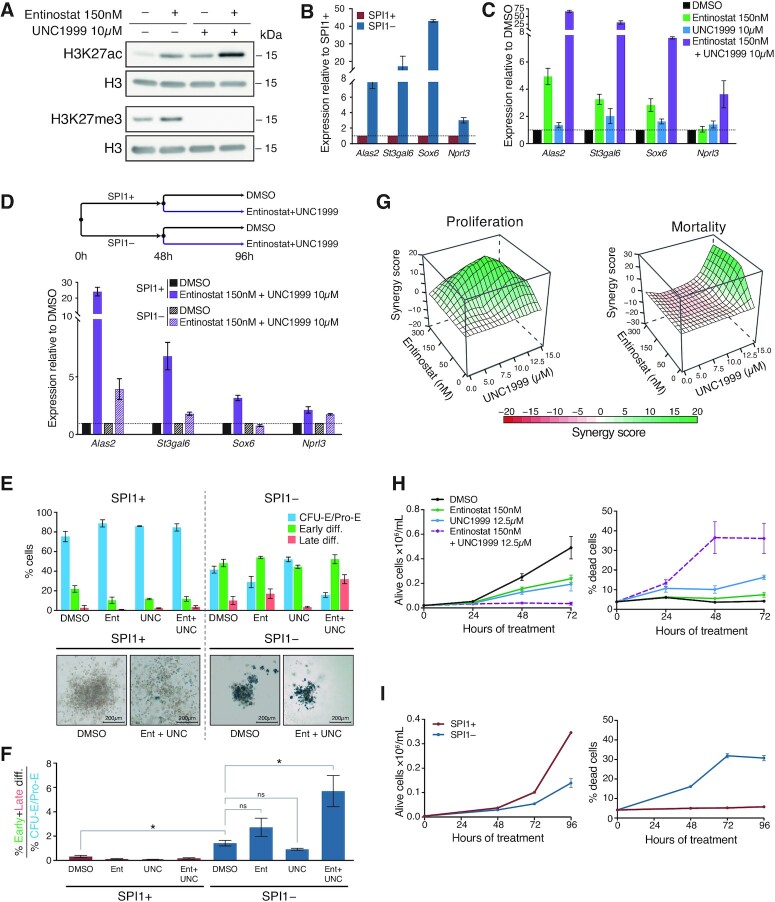
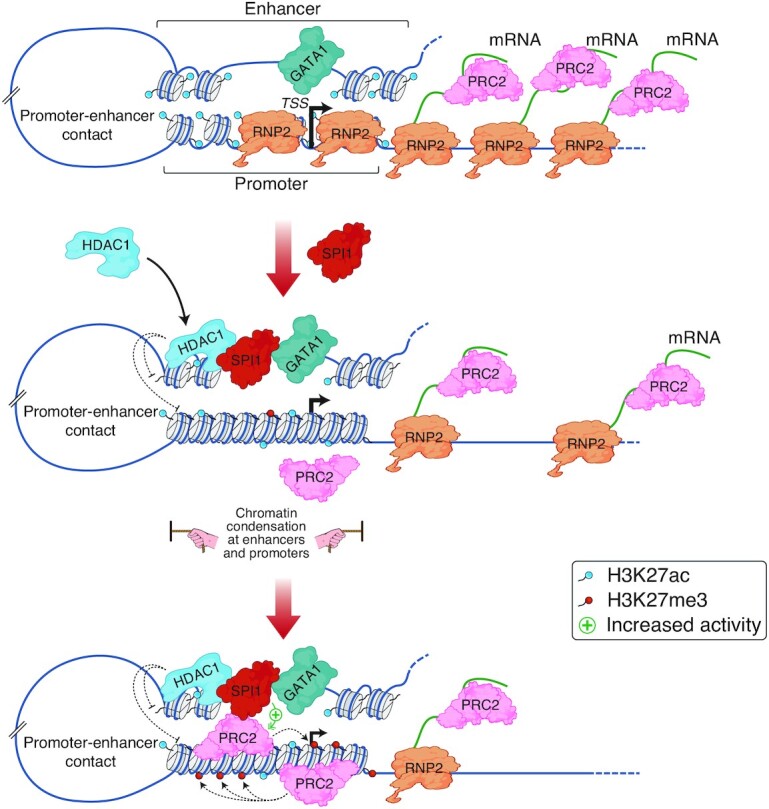
Although originally described as transcriptional activator, SPI1/PU.1, a major player in haematopoiesis whose alterations are associated with haematological malignancies, has the ability to repress transcription. Here, we investigated the mechanisms underlying gene repression in the erythroid lineage, in which SPI1 exerts an oncogenic function by blocking differentiation. We show that SPI1 represses genes by binding active enhancers that are located in intergenic or gene body regions. HDAC1 acts as a cooperative mediator of SPI1-induced transcriptional repression by deacetylating SPI1-bound enhancers in a subset of genes, including those involved in erythroid differentiation. Enhancer deacetylation impacts on promoter acetylation, chromatin accessibility and RNA pol II occupancy. In addition to the activities of HDAC1, polycomb repressive complex 2 (PRC2) reinforces gene repression by depositing H3K27me3 at promoter sequences when SPI1 is located at enhancer sequences. Moreover, our study identified a synergistic relationship between PRC2 and HDAC1 complexes in mediating the transcriptional repression activity of SPI1, ultimately inducing synergistic adverse effects on leukaemic cell survival. Our results highlight the importance of the mechanism underlying transcriptional repression in leukemic cells, involving complex functional connections between SPI1 and the epigenetic regulators PRC2 and HDAC1.
© The Author(s) 2022. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures

References
-
- Kamath M.B., Houston I.B., Janovski A.J., Zhu X., Gowrisankar S., Jegga A.G., DeKoter R.P.. Dose-dependent repression of T-cell and natural killer cell genes by PU.1 enforces myeloid and B-cell identity. Leukemia. 2008; 22:1214–1225. - PubMed
-
- Scott E.W., Simon M.C., Anastasi J., Singh H.. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994; 265:1573–1577. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
