

Prostaglandin F2α and angiotensin II type 1 receptors exhibit differential cognate G protein coupling regulation
- PMID: 35872018
- PMCID: PMC9418914
- DOI: 10.1016/j.jbc.2022.102294
Prostaglandin F2α and angiotensin II type 1 receptors exhibit differential cognate G protein coupling regulation
Abstract
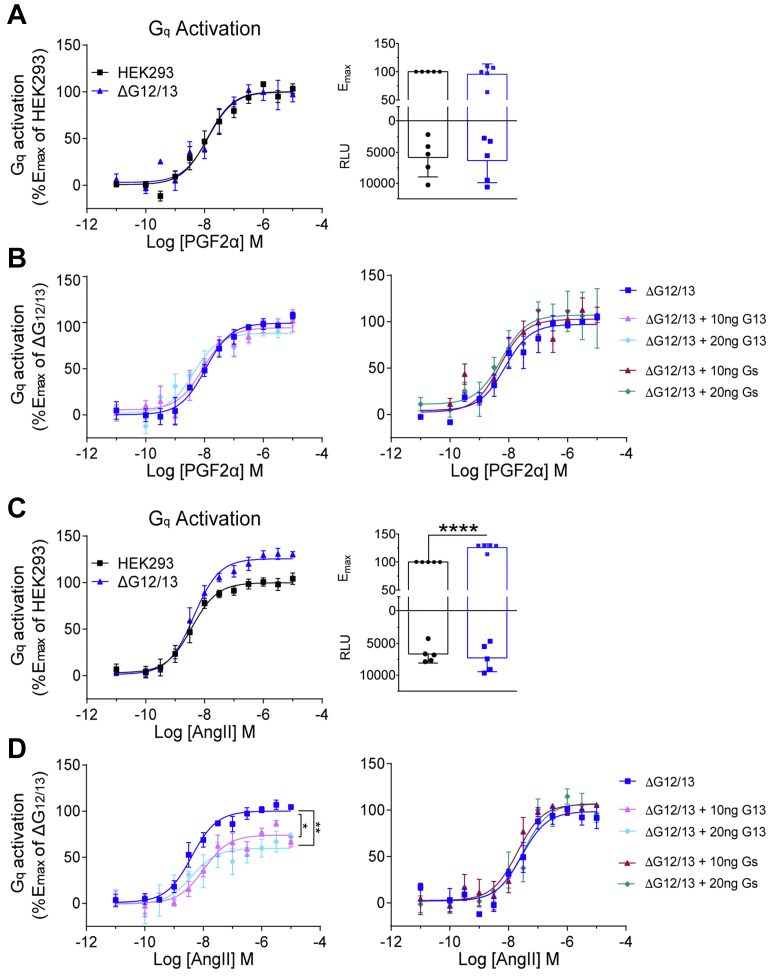
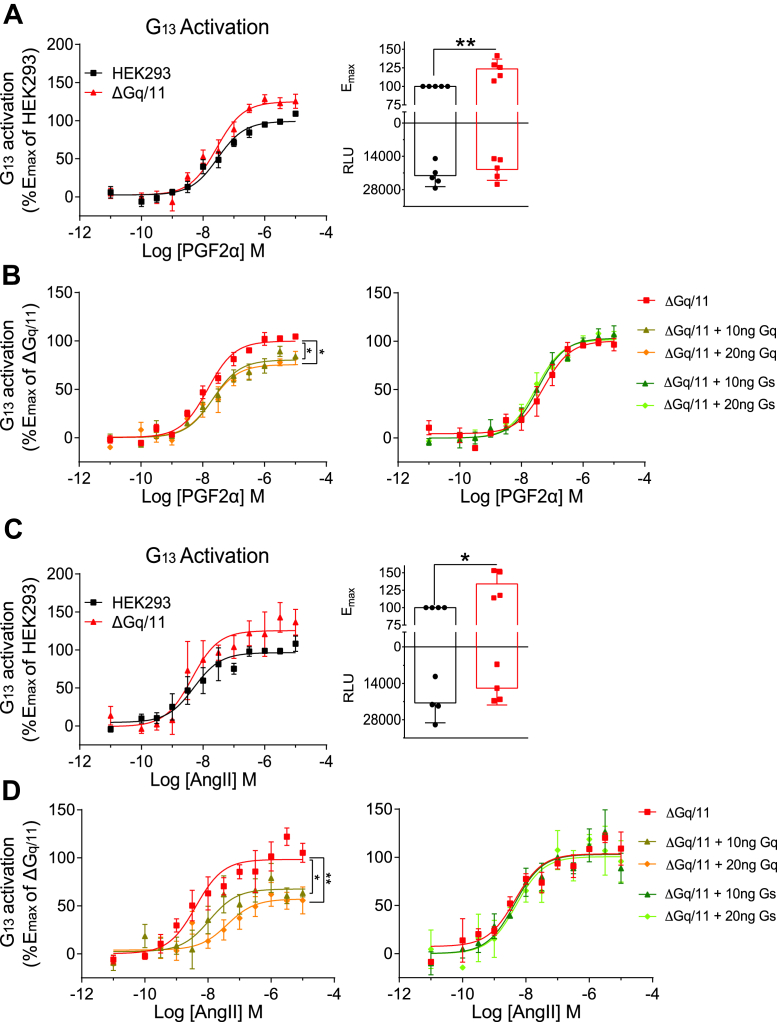
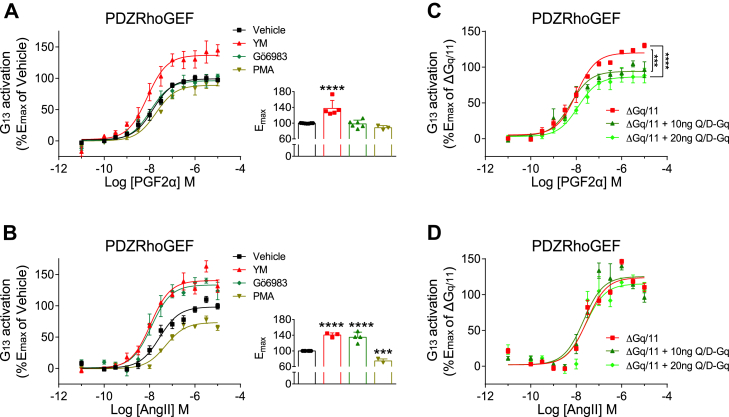
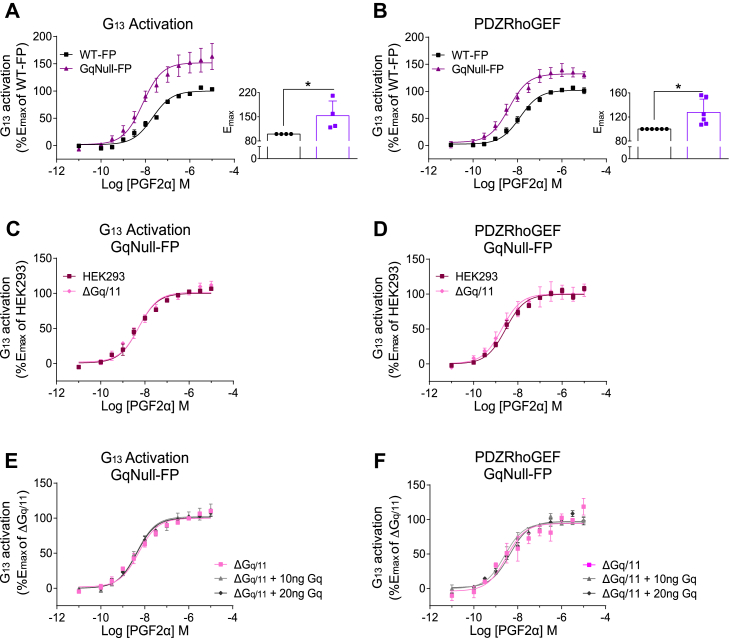
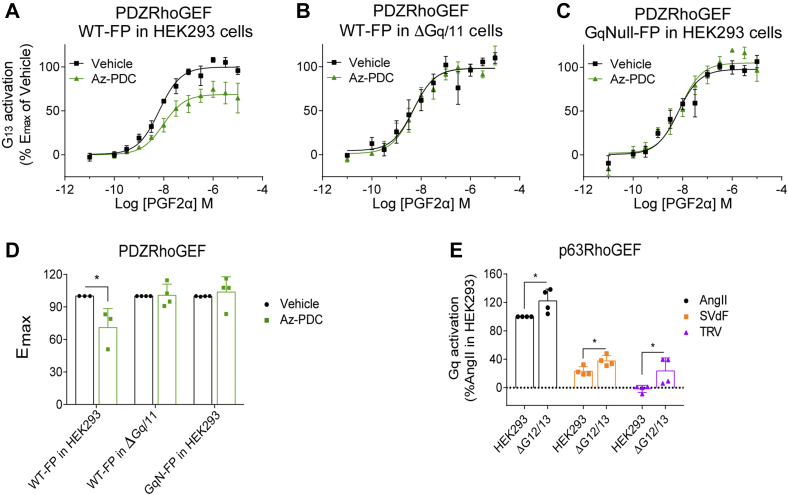
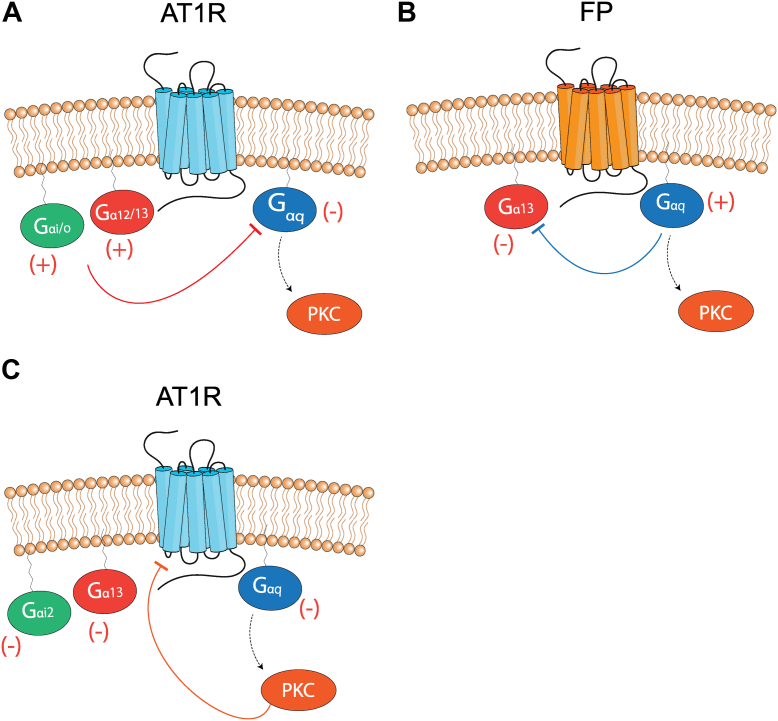
Promiscuous G protein-coupled receptors (GPCRs) engage multiple Gα subtypes with different efficacies to propagate signals in cells. A mechanistic understanding of Gα selectivity by GPCRs is critical for therapeutic design, since signaling can be restrained by ligand-receptor complexes to preferentially engage specific G proteins. However, details of GPCR selectivity are unresolved. Here, we investigated cognate G protein selectivity using the prototypical promiscuous Gαq/11 and Gα12/13 coupling receptors, angiotensin II type I receptor (AT1R) and prostaglandin F2α receptor (FP), bioluminescence resonance energy transfer-based G protein and pathway-selective sensors, and G protein knockout cells. We determined that competition between G proteins for receptor binding occurred in a receptor- and G protein-specific manner for AT1R and FP but not for other receptors tested. In addition, we show that while Gα12/13 competes with Gαq/11 for AT1R coupling, the opposite occurs for FP, and Gαq-mediated signaling regulated G protein coupling only at AT1R. In cells, the functional modulation of biased ligands at FP and AT1R was contingent upon cognate Gα availability. The efficacy of AT1R-biased ligands, which poorly signal through Gαq/11, increased in the absence of Gα12/13. Finally, we show that a positive allosteric modulator of Gαq/11 signaling that also allosterically decreases FP-Gα12/13 coupling, lost its negative modulation in the absence of Gαq/11 coupling to FP. Together, our findings suggest that despite preferential binding of similar subsets of G proteins, GPCRs follow distinct selectivity rules, which may contribute to the regulation of ligand-mediated G protein bias of AT1R and FP.
Keywords: BRET; G protein–coupled receptor; angiotensin II type I receptor; prostaglandin F2α receptor; signaling.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest Some of the BRET biosensors used in the present study are licensed to Domain Therapeutics for commercial use. The biosensors are freely available under material transfer agreement for academic research and can be requested from S. A. L. All other authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
Conformational biosensors reveal allosteric interactions between heterodimeric AT1 angiotensin and prostaglandin F2α receptors.J Biol Chem. 2017 Jul 21;292(29):12139-12152. doi: 10.1074/jbc.M117.793877. Epub 2017 Jun 5. J Biol Chem. 2017. PMID: 28584054 Free PMC article.
-
Angiotensin II type I and prostaglandin F2α receptors cooperatively modulate signaling in vascular smooth muscle cells.J Biol Chem. 2015 Jan 30;290(5):3137-48. doi: 10.1074/jbc.M114.631119. Epub 2014 Dec 15. J Biol Chem. 2015. PMID: 25512374 Free PMC article.
-
Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors.Sci Signal. 2018 Dec 4;11(559):eaat1631. doi: 10.1126/scisignal.aat1631. Sci Signal. 2018. PMID: 30514808
-
Cardiovascular angiotensin II type 1 receptor biased signaling: Focus on non-Gq-, non-βarrestin-dependent signaling.Pharmacol Res. 2021 Dec;174:105943. doi: 10.1016/j.phrs.2021.105943. Epub 2021 Oct 15. Pharmacol Res. 2021. PMID: 34662735 Review.
-
Biased agonism of the angiotensin II type 1 receptor.Mini Rev Med Chem. 2012 Aug;12(9):812-6. doi: 10.2174/138955712800959134. Mini Rev Med Chem. 2012. PMID: 22681254 Review.
Cited by
-
G12/13 signaling in asthma.Respir Res. 2024 Aug 2;25(1):295. doi: 10.1186/s12931-024-02920-0. Respir Res. 2024. PMID: 39095798 Free PMC article. Review.
-
Optimization of the prostaglandin F2α receptor for structural biology.PLoS One. 2025 Jul 18;20(7):e0320114. doi: 10.1371/journal.pone.0320114. eCollection 2025. PLoS One. 2025. PMID: 40680049 Free PMC article.
-
Unraveling allostery within the angiotensin II type 1 receptor for Gαq and β-arrestin coupling.Sci Signal. 2023 Aug 8;16(797):eadf2173. doi: 10.1126/scisignal.adf2173. Epub 2023 Aug 8. Sci Signal. 2023. PMID: 37552769 Free PMC article.
References
-
- Wettschureck N., Offermanns S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005;85:1159–1204. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
