

Challenges in the Treatment of Glioblastoma by Chimeric Antigen Receptor T-Cell Immunotherapy and Possible Solutions
- PMID: 35874698
- PMCID: PMC9300859
- DOI: 10.3389/fimmu.2022.927132
Challenges in the Treatment of Glioblastoma by Chimeric Antigen Receptor T-Cell Immunotherapy and Possible Solutions
Abstract
Glioblastoma (GBM), one of the most lethal brain cancers in adults, accounts for 48.6% of all malignant primary CNS tumors diagnosed each year. The 5-year survival rate of GBM patients remains less than 10% even after they receive the standard-of-care treatment, including maximal safe resection, adjuvant radiation, and chemotherapy with temozolomide. Therefore, new therapeutic modalities are urgently needed for this deadly cancer. The last decade has witnessed great advances in chimeric antigen receptor T (CAR-T) cell immunotherapy for the treatment of hematological malignancies. Up to now, the US FDA has approved six CAR-T cell products in treating hematopoietic cancers including B-cell acute lymphoblastic leukemia, lymphoma, and multiple myeloma. Meanwhile, the number of clinical trials on CAR-T cell has increased significantly, with more than 80% from China and the United States. With its achievements in liquid cancers, the clinical efficacy of CAR-T cell therapy has also been explored in a variety of solid malignancies that include GBMs. However, attempts to expand CAR-T cell immunotherapy in GBMs have not yet presented promising results in hematopoietic malignancies. Like other solid tumors, CAR-T cell therapies against GBM still face several challenges, such as tumor heterogeneity, tumor immunosuppressive microenvironment, and CAR-T cell persistence. Hence, developing strategies to overcome these challenges will be necessary to accelerate the transition of CAR-T cell immunotherapy against GBMs from bench to bedside.
Keywords: CAR-T; adoptive immunotherapy; cellular immunotherapy; chimeric antigen receptor; glioblastoma.
Copyright © 2022 Zhang, Zhang and Ji.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
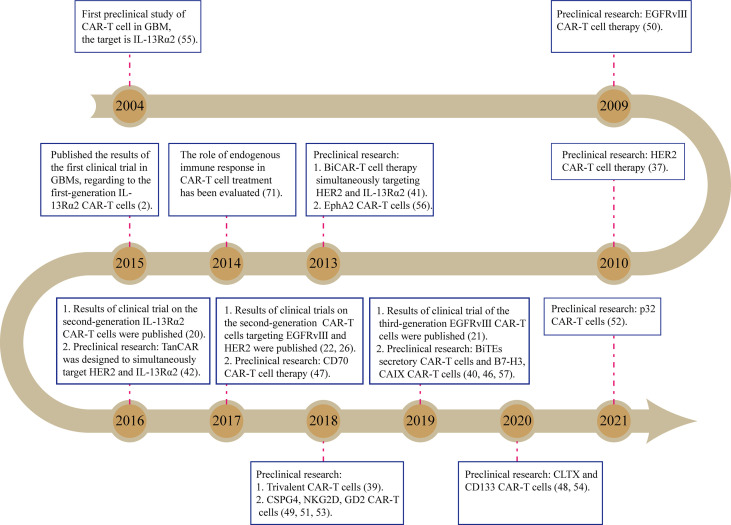
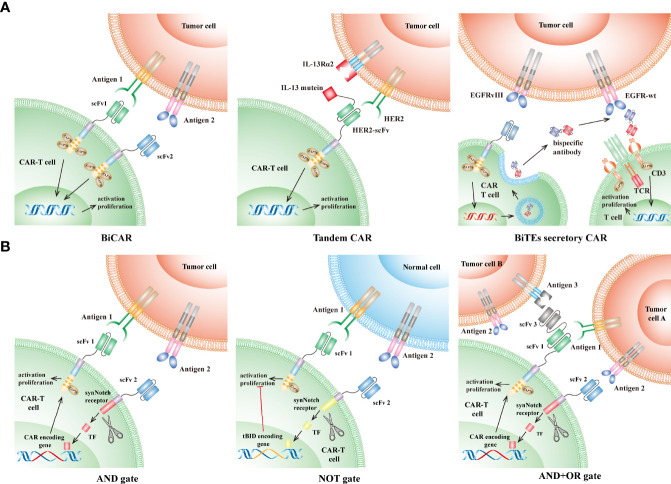


References
-
- Sadelain M, Brentjens R, Riviere I. The Basic Principles of Chimeric Antigen Receptor Design. Cancer Discov (2013) 3(4):388–98. doi: 10.1158/2159-8290.CD-12-0548 - DOI - PMC - PubMed
-
- Brown CE, Starr R, Aguilar B, Shami AF, Martinez C, D'Apuzzo M, et al. Stem-Like Tumor-Initiating Cells Isolated From IL13Ralpha2 Expressing Gliomas Are Targeted and Killed by IL13-Zetakine-Redirected T Cells. Clin Cancer Res (2012) 18(8):2199–209. doi: 10.1158/1078-0432.CCR-11-1669 - DOI - PMC - PubMed
-
- Ohno M, Natsume A, Ichiro Iwami K, Iwamizu H, Noritake K, Ito D, et al. Retrovirally Engineered T-Cell-Based Immunotherapy Targeting Type III Variant Epidermal Growth Factor Receptor, a Glioma-Associated Antigen. Cancer Sci (2010) 101(12):2518–24. doi: 10.1111/j.1349-7006.2010.01734.x - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources