

Protein Science Meets Artificial Intelligence: A Systematic Review and a Biochemical Meta-Analysis of an Inter-Field
- PMID: 35875501
- PMCID: PMC9301016
- DOI: 10.3389/fbioe.2022.788300
Protein Science Meets Artificial Intelligence: A Systematic Review and a Biochemical Meta-Analysis of an Inter-Field
Abstract
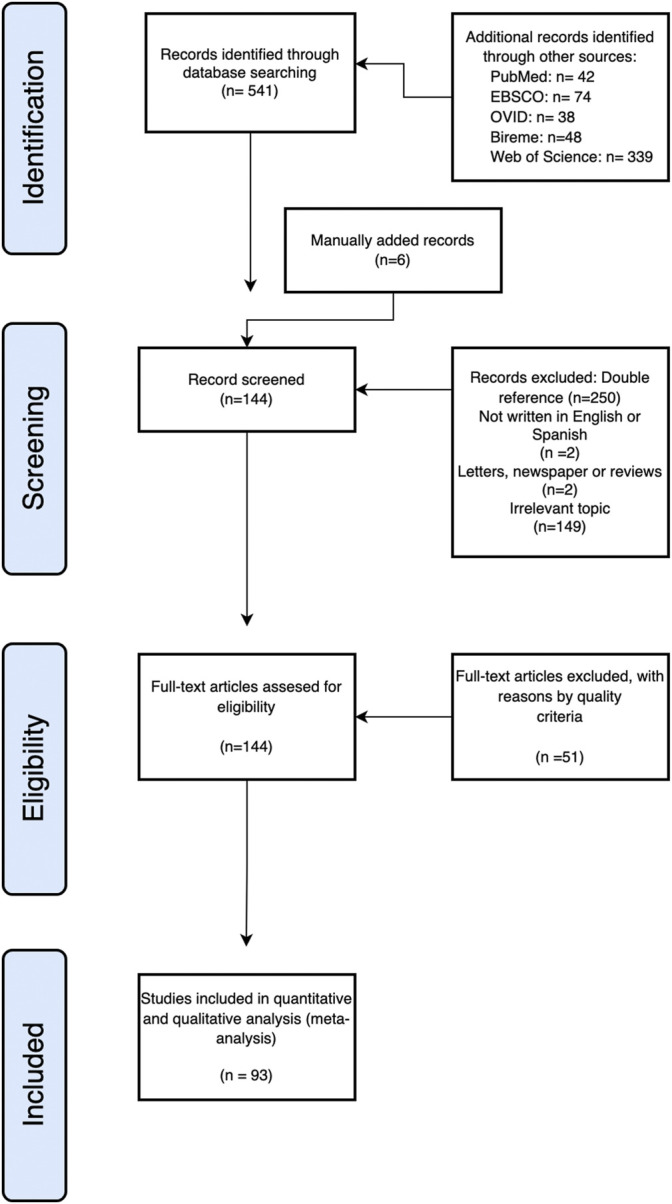
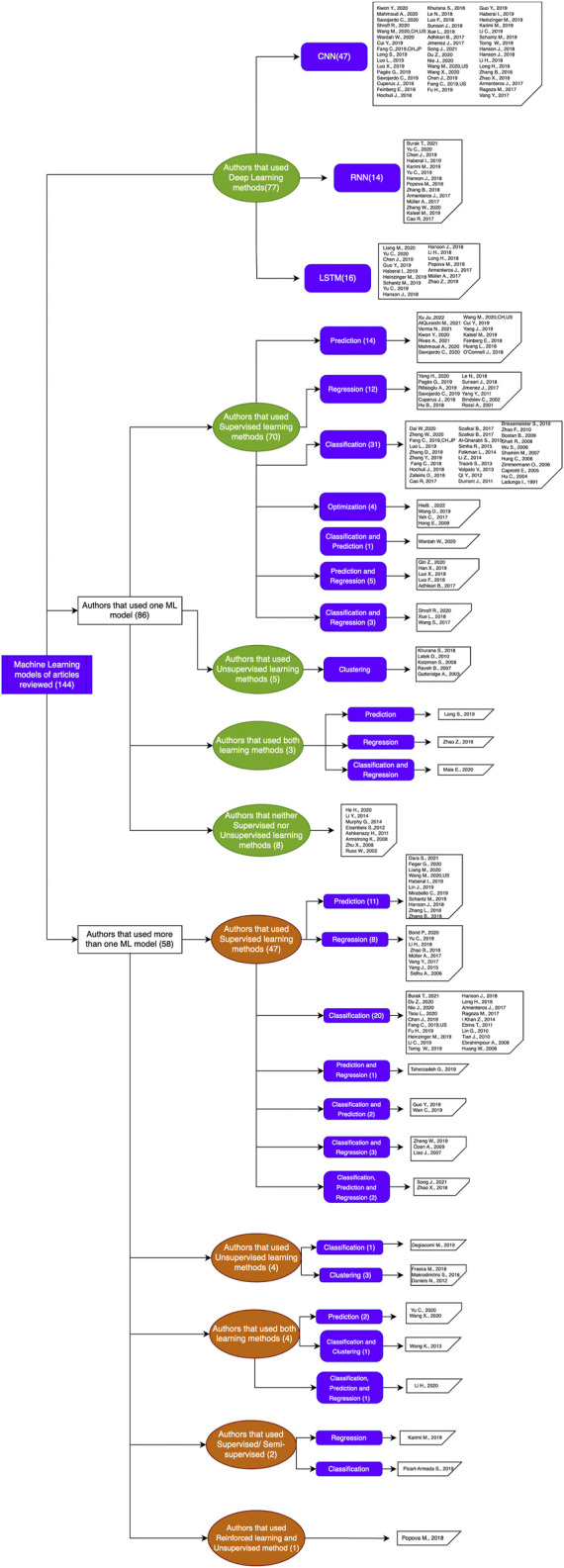
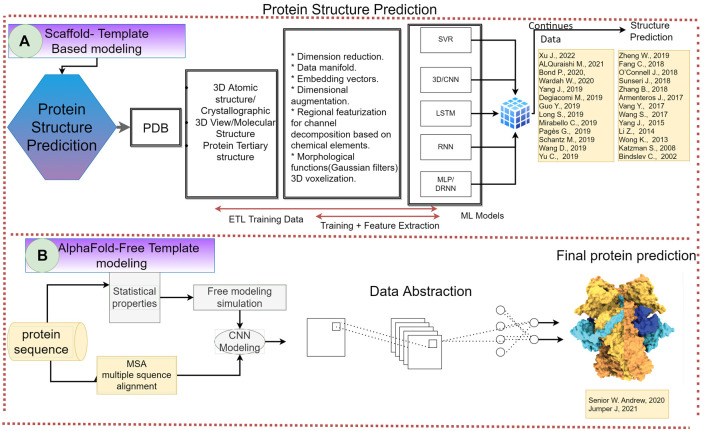
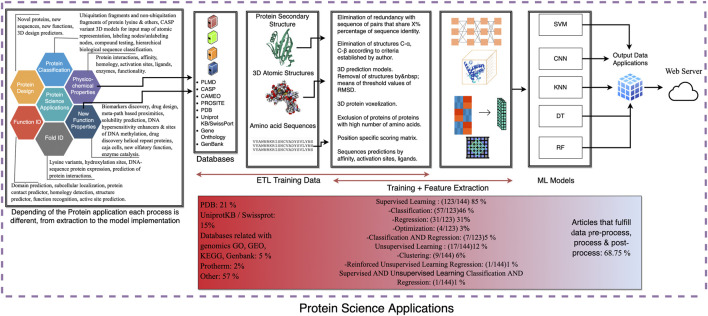
Proteins are some of the most fascinating and challenging molecules in the universe, and they pose a big challenge for artificial intelligence. The implementation of machine learning/AI in protein science gives rise to a world of knowledge adventures in the workhorse of the cell and proteome homeostasis, which are essential for making life possible. This opens up epistemic horizons thanks to a coupling of human tacit-explicit knowledge with machine learning power, the benefits of which are already tangible, such as important advances in protein structure prediction. Moreover, the driving force behind the protein processes of self-organization, adjustment, and fitness requires a space corresponding to gigabytes of life data in its order of magnitude. There are many tasks such as novel protein design, protein folding pathways, and synthetic metabolic routes, as well as protein-aggregation mechanisms, pathogenesis of protein misfolding and disease, and proteostasis networks that are currently unexplored or unrevealed. In this systematic review and biochemical meta-analysis, we aim to contribute to bridging the gap between what we call binomial artificial intelligence (AI) and protein science (PS), a growing research enterprise with exciting and promising biotechnological and biomedical applications. We undertake our task by exploring "the state of the art" in AI and machine learning (ML) applications to protein science in the scientific literature to address some critical research questions in this domain, including What kind of tasks are already explored by ML approaches to protein sciences? What are the most common ML algorithms and databases used? What is the situational diagnostic of the AI-PS inter-field? What do ML processing steps have in common? We also formulate novel questions such as Is it possible to discover what the rules of protein evolution are with the binomial AI-PS? How do protein folding pathways evolve? What are the rules that dictate the folds? What are the minimal nuclear protein structures? How do protein aggregates form and why do they exhibit different toxicities? What are the structural properties of amyloid proteins? How can we design an effective proteostasis network to deal with misfolded proteins? We are a cross-functional group of scientists from several academic disciplines, and we have conducted the systematic review using a variant of the PICO and PRISMA approaches. The search was carried out in four databases (PubMed, Bireme, OVID, and EBSCO Web of Science), resulting in 144 research articles. After three rounds of quality screening, 93 articles were finally selected for further analysis. A summary of our findings is as follows: regarding AI applications, there are mainly four types: 1) genomics, 2) protein structure and function, 3) protein design and evolution, and 4) drug design. In terms of the ML algorithms and databases used, supervised learning was the most common approach (85%). As for the databases used for the ML models, PDB and UniprotKB/Swissprot were the most common ones (21 and 8%, respectively). Moreover, we identified that approximately 63% of the articles organized their results into three steps, which we labeled pre-process, process, and post-process. A few studies combined data from several databases or created their own databases after the pre-process. Our main finding is that, as of today, there are no research road maps serving as guides to address gaps in our knowledge of the AI-PS binomial. All research efforts to collect, integrate multidimensional data features, and then analyze and validate them are, so far, uncoordinated and scattered throughout the scientific literature without a clear epistemic goal or connection between the studies. Therefore, our main contribution to the scientific literature is to offer a road map to help solve problems in drug design, protein structures, design, and function prediction while also presenting the "state of the art" on research in the AI-PS binomial until February 2021. Thus, we pave the way toward future advances in the synthetic redesign of novel proteins and protein networks and artificial metabolic pathways, learning lessons from nature for the welfare of humankind. Many of the novel proteins and metabolic pathways are currently non-existent in nature, nor are they used in the chemical industry or biomedical field.
Keywords: artificial intelligence; deep learning; drug design; machine learning; protein classification; protein design and engineering; protein prediction; proteins.
Copyright © 2022 Villalobos-Alva, Ochoa-Toledo, Villalobos-Alva, Aliseda, Pérez-Escamirosa, Altamirano-Bustamante, Ochoa-Fernández, Zamora-Solís, Villalobos-Alva, Revilla-Monsalve, Kemper-Valverde and Altamirano-Bustamante.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures

Similar articles
-
Artificial intelligence to deep learning: machine intelligence approach for drug discovery.Mol Divers. 2021 Aug;25(3):1315-1360. doi: 10.1007/s11030-021-10217-3. Epub 2021 Apr 12. Mol Divers. 2021. PMID: 33844136 Free PMC article. Review.
-
Embryo selection through artificial intelligence versus embryologists: a systematic review.Hum Reprod Open. 2023 Aug 15;2023(3):hoad031. doi: 10.1093/hropen/hoad031. eCollection 2023. Hum Reprod Open. 2023. PMID: 37588797 Free PMC article.
-
Beginnings of Artificial Intelligence in Medicine (AIM): Computational Artifice Assisting Scientific Inquiry and Clinical Art - with Reflections on Present AIM Challenges.Yearb Med Inform. 2019 Aug;28(1):249-256. doi: 10.1055/s-0039-1677895. Epub 2019 Apr 25. Yearb Med Inform. 2019. PMID: 31022744 Free PMC article.
-
The future of Cochrane Neonatal.Early Hum Dev. 2020 Nov;150:105191. doi: 10.1016/j.earlhumdev.2020.105191. Epub 2020 Sep 12. Early Hum Dev. 2020. PMID: 33036834
-
Data-driven modeling and prediction of blood glucose dynamics: Machine learning applications in type 1 diabetes.Artif Intell Med. 2019 Jul;98:109-134. doi: 10.1016/j.artmed.2019.07.007. Epub 2019 Jul 26. Artif Intell Med. 2019. PMID: 31383477 Review.
Cited by
-
Artificial intelligence driven innovations in biochemistry: A review of emerging research frontiers.Biomol Biomed. 2025 Mar 7;25(4):739-750. doi: 10.17305/bb.2024.11537. Biomol Biomed. 2025. PMID: 39819459 Free PMC article. Review.
-
Int&in: A machine learning-based web server for active split site identification in inteins.Protein Sci. 2024 Jun;33(6):e4985. doi: 10.1002/pro.4985. Protein Sci. 2024. PMID: 38717278 Free PMC article.
References
-
- Alakuş T. B., Türkoğlu İ. (2021). A Novel Fibonacci Hash Method for Protein Family Identification by Using Recurrent Neural Networks. Turk. J. Electr. Eng. Comput. Sci. 29, 370–386. Available at: http://10.0.15.66/elk-2003-116 . 10.0.15.66/elk-2003-116 - DOI - DOI
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous