

Enhancement of Proteome Coverage by Ion Mobility Fractionation Coupled to PASEF on a TIMS-QTOF Instrument
- PMID: 35876248
- PMCID: PMC10653119
- DOI: 10.1021/acs.jproteome.2c00336
Enhancement of Proteome Coverage by Ion Mobility Fractionation Coupled to PASEF on a TIMS-QTOF Instrument
Abstract
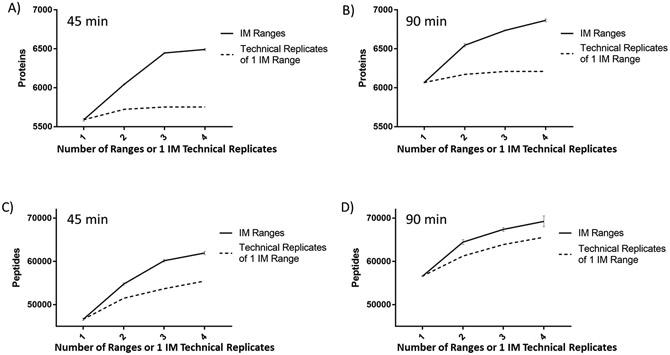
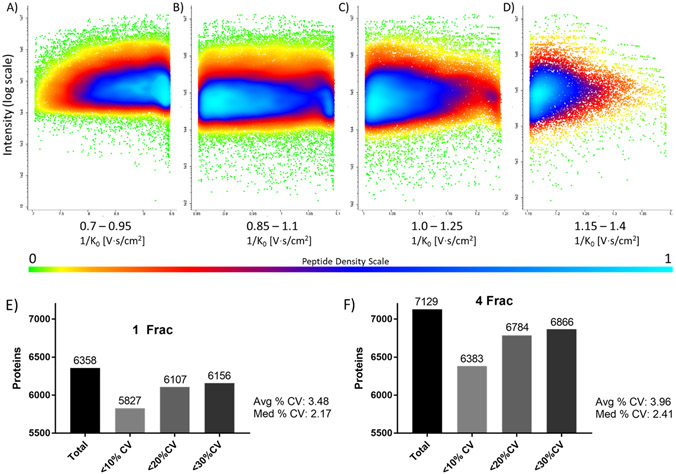
Trapped ion-mobility spectrometry (TIMS) was used to fractionate ions in the gas phase based on their ion mobility (V s/cm2), followed by parallel accumulation-serial fragmentation (PASEF) using a quadrupole time-of-flight instrument to determine the effect on the depth of proteome coverage. TIMS fractionation (up to four gas-phase fractions) coupled to data-dependent acquisition (DDA)-PASEF resulted in the detection of ∼7000 proteins and over 70,000 peptides overall from 200 ng of human (HeLa) cell lysate per injection using a commercial 25 cm ultra high performance liquid chromatography (UHPLC) column with a 90 min gradient. This result corresponded to ∼19 and 30% increases in protein and peptide identifications, respectively, when compared to a default, single-range TIMS DDA-PASEF analysis. Quantitation precision was not affected by TIMS fractionation as demonstrated by the average and median coefficient of variation values that were less than 4% upon label-free quantitation of technical replicates. TIMS fractionation was utilized to generate a DDA-based spectral library for downstream data-independent acquisition (DIA) analysis of lower sample input using a shorter LC gradient. The TIMS-fractionated library, consisting of over 7600 proteins and 82,000 peptides, enabled the identification of ∼4000 and 6600 proteins from 10 and 200 ng of human (HeLa) cell lysate input, respectively, with a 20 min gradient, single-shot DIA analysis. Data are available in ProteomeXchange: identifier PXD033129.
Keywords: DDA-PASEF; DIA-PASEF; gas-phase fractionation; ion mobility; proteomics; spectral library; timsTOF Pro.
Figures




References
-
- Link AJ; Eng J; Schieltz DM; Carmack E; Mize GJ; Morris DR; Garvik BM; Yates JR 3rd, Direct analysis of protein complexes using mass spectrometry. Nat Biotechnol 1999, 17 (7), 676–82. - PubMed
-
- Washburn MP; Wolters D; Yates JR 3rd, Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol 2001, 19 (3), 242–7. - PubMed
-
- Spahr CS; Davis MT; McGinley MD; Robinson JH; Bures EJ; Beierle J; Mort J; Courchesne PL; Chen K; Wahl RC; Yu W; Luethy R; Patterson SD, Towards defining the urinary proteome using liquid chromatography-tandem mass spectrometry. I. Profiling an unfractionated tryptic digest. Proteomics 2001, 1 (1), 93–107. - PubMed
-
- Yi EC; Marelli M; Lee H; Purvine SO; Aebersold R; Aitchison JD; Goodlett DR, Approaching complete peroxisome characterization by gas-phase fractionation. Electrophoresis 2002, 23 (18), 3205–16. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
