

Immunopathogenesis and environmental triggers in coeliac disease
- PMID: 35879049
- PMCID: PMC9554150
- DOI: 10.1136/gutjnl-2021-326257
Immunopathogenesis and environmental triggers in coeliac disease
Abstract
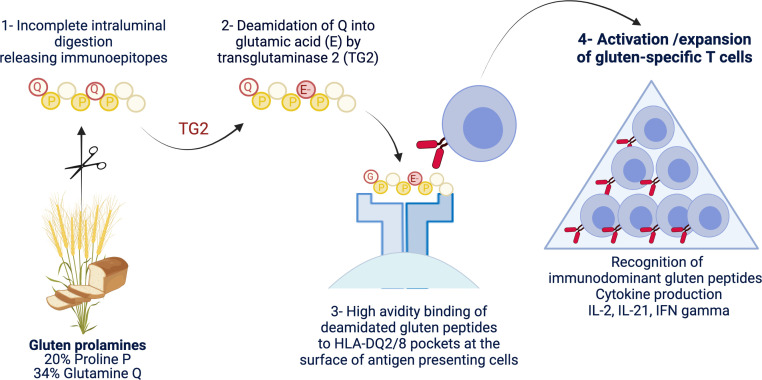
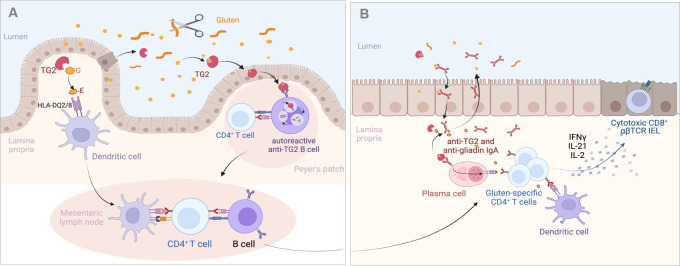
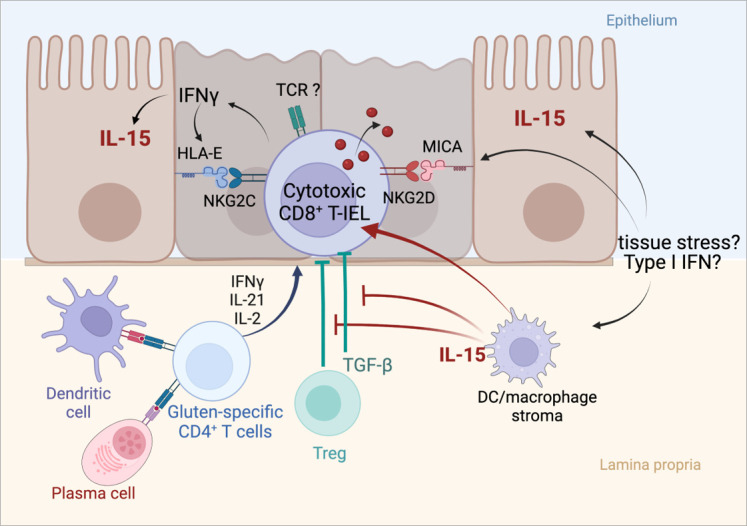
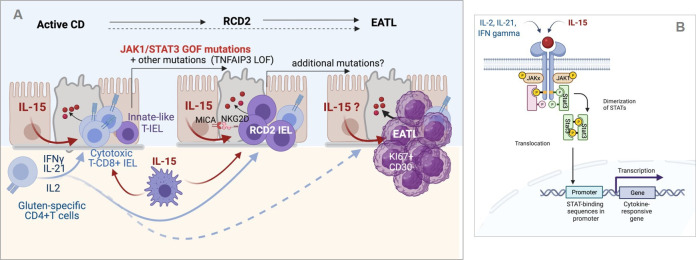
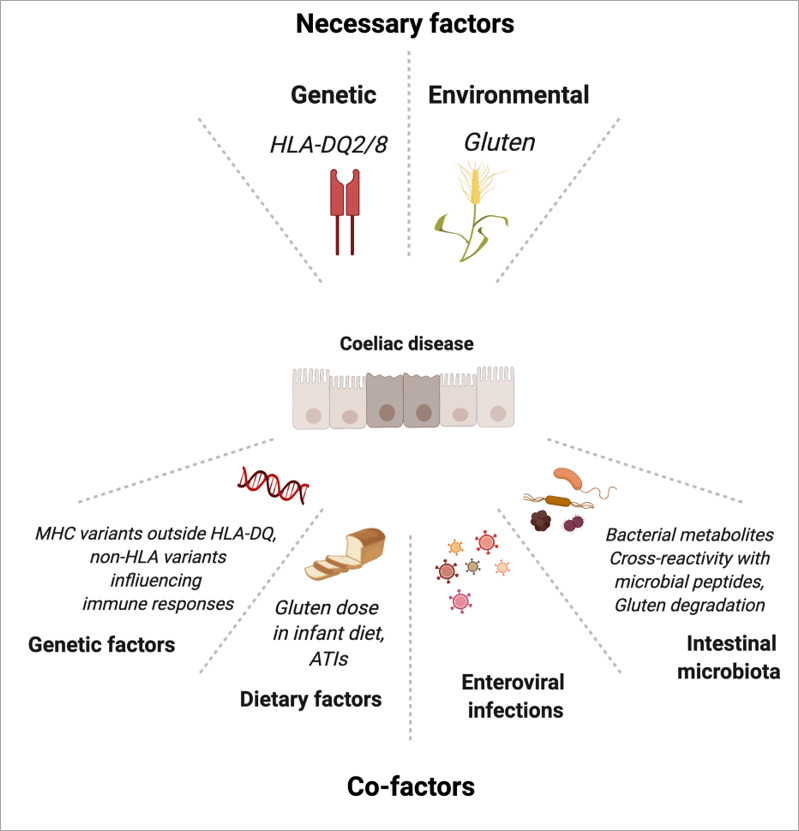
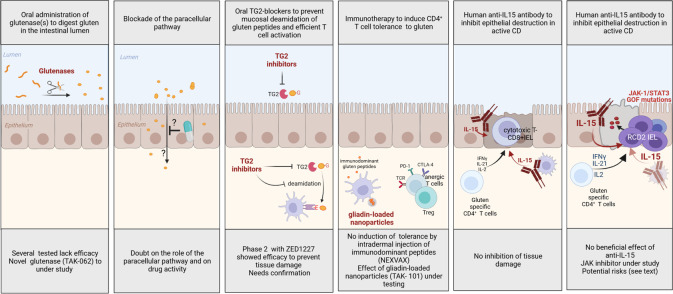
Coeliac disease (CD) is a frequent immune enteropathy induced by gluten in genetically predisposed individuals. Its pathogenesis has been extensively studied and CD has emerged as a model disease to decipher how the interplay between environmental and genetic factors can predispose to autoimmunity and promote lymphomagenesis. The keystone event is the activation of a gluten-specific immune response that is driven by molecular interactions between gluten, the indispensable environmental factor, HLA-DQ2/8, the main predisposing genetic factor and transglutaminase 2, the CD-specific autoantigen. The antigluten response is however not sufficient to induce epithelial damage which requires the activation of cytotoxic CD8+ intraepithelial lymphocytes (IEL). In a plausible scenario, cooperation between cytokines released by gluten-specific CD4+ T cells and interleukin-15 produced in excess in the coeliac gut, licenses the autoimmune-like attack of the gut epithelium, likely via sustained activation of the Janus kinase-signal transducer and activator of transcription (JAK/STAT) pathway in IEL. Demonstration that lymphomas complicating CD arise from IEL that have acquired gain-of-function JAK1 or STAT3 mutations stresses the key role of this pathway and explains how gluten-driven chronic inflammation may promote this rare but most severe complication. If our understanding of CD pathogenesis has considerably progressed, several questions and challenges remain. One unsolved question concerns the considerable variability in disease penetrance, severity and presentation, pointing to the role of additional genetic and environmental factors that remain however uneasy to untangle and hierarchize. A current challenge is to transfer the considerable mechanistic insight gained into CD pathogenesis into benefits for the patients, notably to alleviate the gluten-free diet, a burden for many patients.
Keywords: T lymphocytes; autoimmunity; coeliac disease; gluten sensitive enteropathy; lymphoma.
© Author(s) (or their employer(s)) 2022. Re-use permitted under CC BY-NC. No commercial re-use. See rights and permissions. Published by BMJ.
Conflict of interest statement
Competing interests: None declared.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous