

Ras-mutant cancers are sensitive to small molecule inhibition of V-type ATPases in mice
- PMID: 35879364
- PMCID: PMC9750872
- DOI: 10.1038/s41587-022-01386-z
Ras-mutant cancers are sensitive to small molecule inhibition of V-type ATPases in mice
Abstract
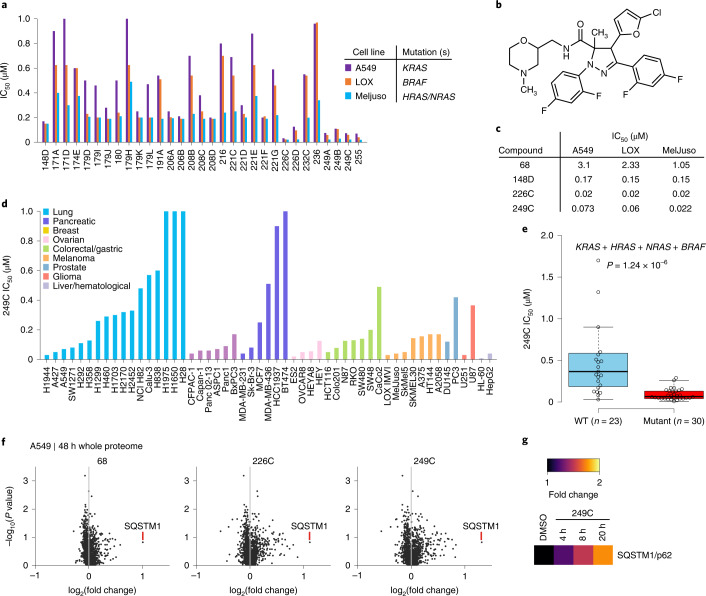
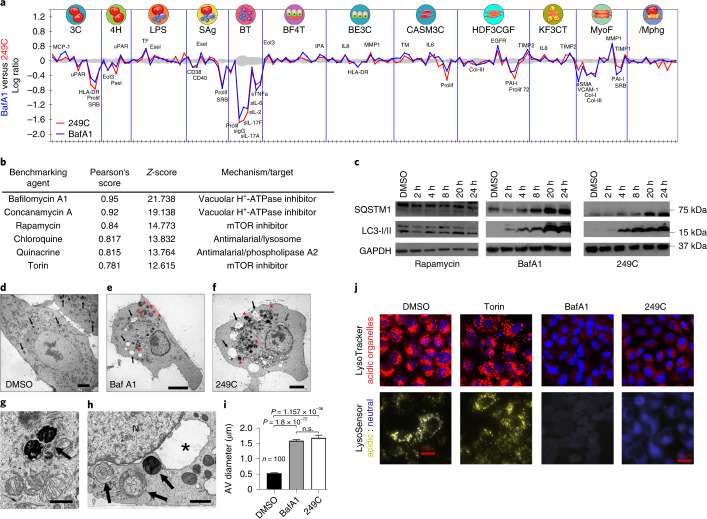
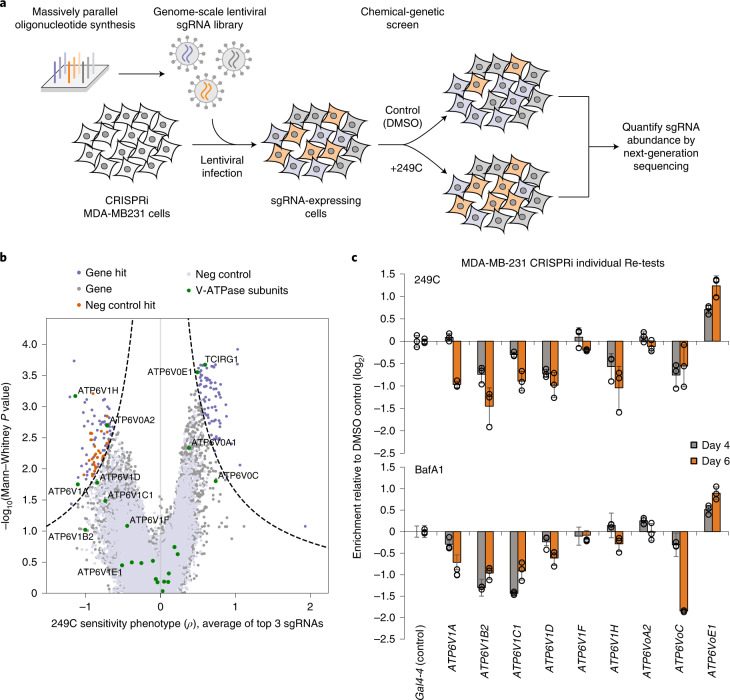
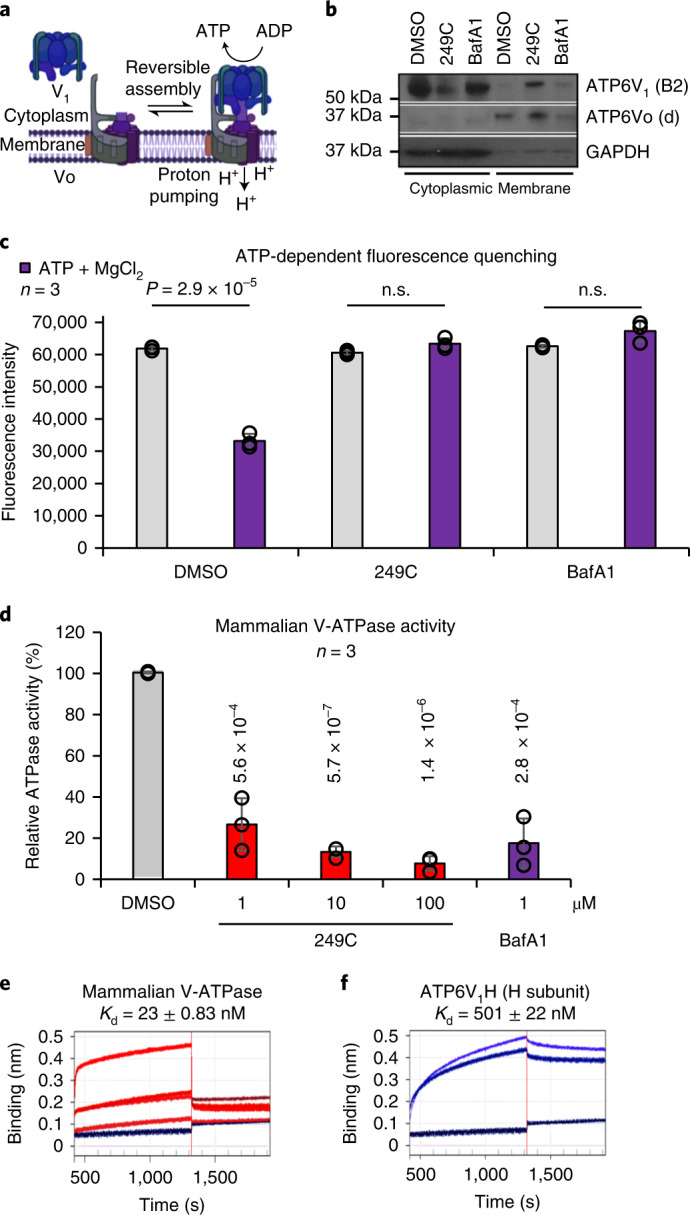
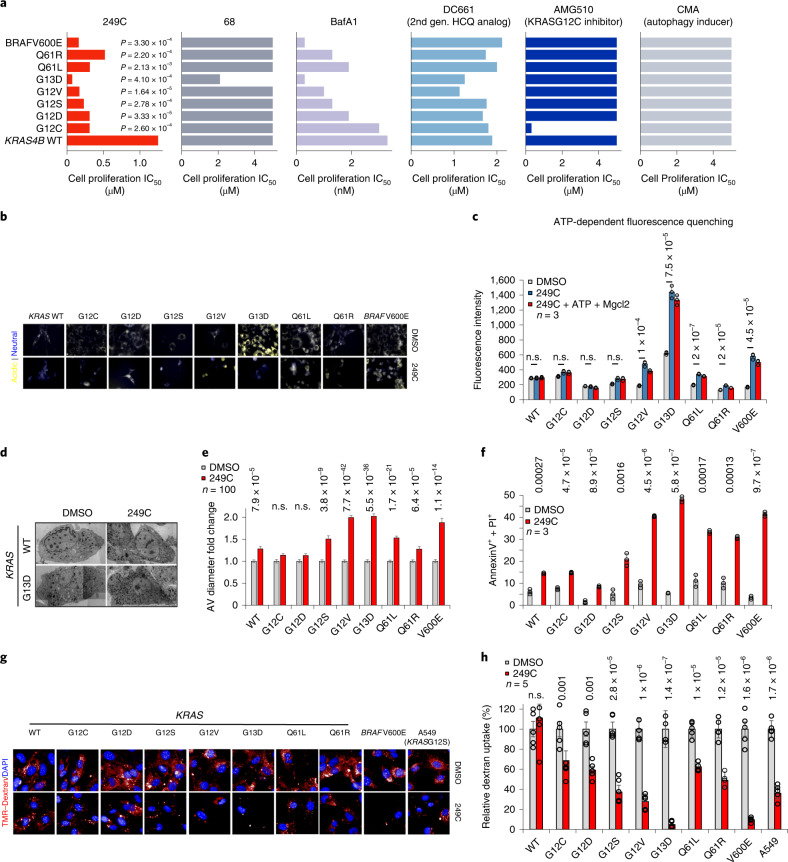
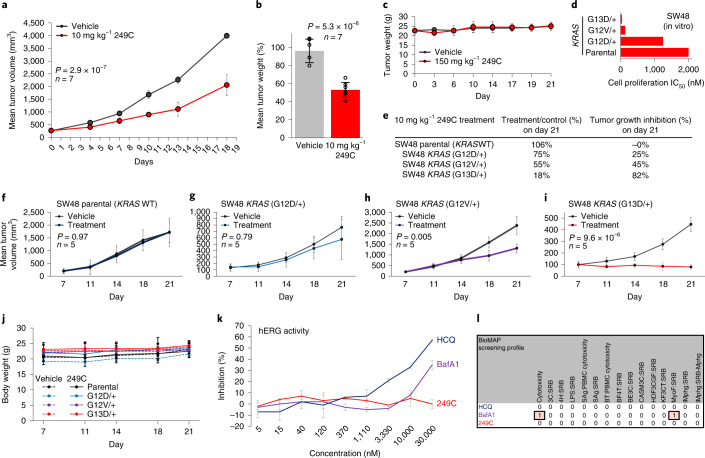
Mutations in Ras family proteins are implicated in 33% of human cancers, but direct pharmacological inhibition of Ras mutants remains challenging. As an alternative to direct inhibition, we screened for sensitivities in Ras-mutant cells and discovered 249C as a Ras-mutant selective cytotoxic agent with nanomolar potency against a spectrum of Ras-mutant cancers. 249C binds to vacuolar (V)-ATPase with nanomolar affinity and inhibits its activity, preventing lysosomal acidification and inhibiting autophagy and macropinocytosis pathways that several Ras-driven cancers rely on for survival. Unexpectedly, potency of 249C varies with the identity of the Ras driver mutation, with the highest potency for KRASG13D and G12V both in vitro and in vivo, highlighting a mutant-specific dependence on macropinocytosis and lysosomal pH. Indeed, 249C potently inhibits tumor growth without adverse side effects in mouse xenografts of KRAS-driven lung and colon cancers. A comparison of isogenic SW48 xenografts with different KRAS mutations confirmed that KRASG13D/+ (followed by G12V/+) mutations are especially sensitive to 249C treatment. These data establish proof-of-concept for targeting V-ATPase in cancers driven by specific KRAS mutations such as KRASG13D and G12V.
© 2022. The Author(s).
Conflict of interest statement
B.T. is an inventor on the patent filed by UCSF covering these molecules. UCSF has licensed this patent to a start-up biotechnology company. The startup calls 249C ‘RSC-1255’ instead and is running a Phase Ia/1b dose escalation and dose expansion clinical trial for the following indications: Ras mutations; lung cancer; colon cancer; pancreatic cancer; and glioblastoma. It is being run as a multi-center trial across five different sites in the U.S. Recruitment is ongoing (
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
