

Mitochondrial control of inflammation
- PMID: 35879417
- PMCID: PMC9310369
- DOI: 10.1038/s41577-022-00760-x
Mitochondrial control of inflammation
Abstract
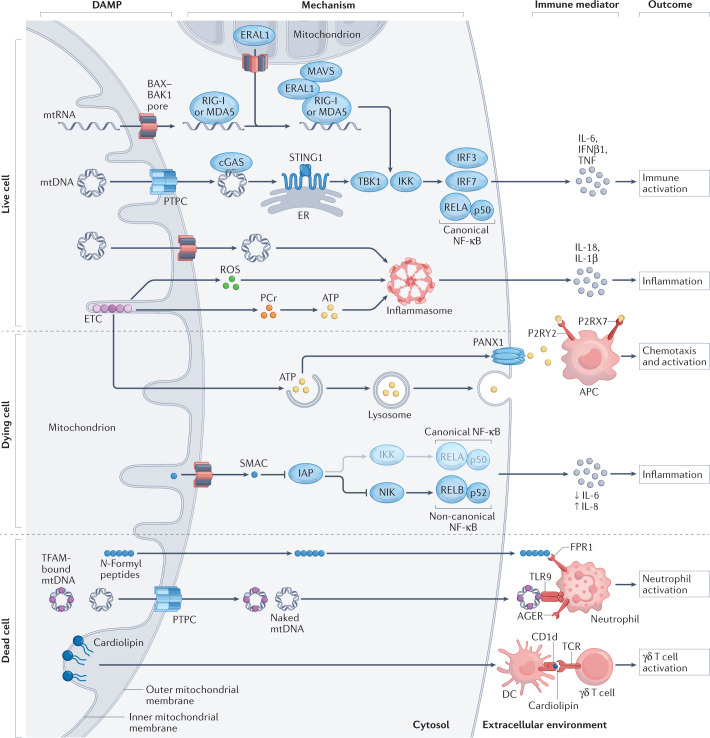
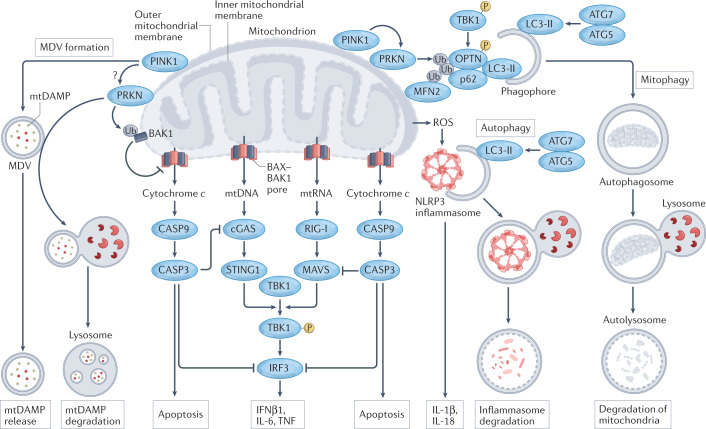
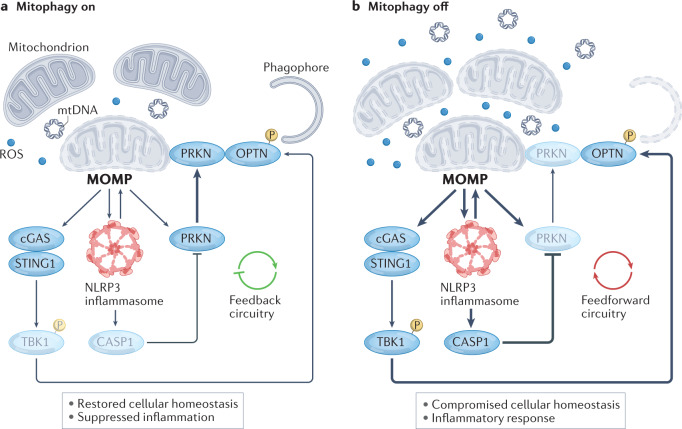
Numerous mitochondrial constituents and metabolic products can function as damage-associated molecular patterns (DAMPs) and promote inflammation when released into the cytosol or extracellular milieu. Several safeguards are normally in place to prevent mitochondria from eliciting detrimental inflammatory reactions, including the autophagic disposal of permeabilized mitochondria. However, when the homeostatic capacity of such systems is exceeded or when such systems are defective, inflammatory reactions elicited by mitochondria can become pathogenic and contribute to the aetiology of human disorders linked to autoreactivity. In addition, inefficient inflammatory pathways induced by mitochondrial DAMPs can be pathogenic as they enable the establishment or progression of infectious and neoplastic disorders. Here we discuss the molecular mechanisms through which mitochondria control inflammatory responses, the cellular pathways that are in place to control mitochondria-driven inflammation and the pathological consequences of dysregulated inflammatory reactions elicited by mitochondrial DAMPs.
© 2022. Springer Nature Limited.
Conflict of interest statement
L.G. has held research contracts with Lytix Biopharma and Promontory, has received consulting/advisory honoraria from Boehringer Ingelheim, AstraZeneca, OmniSEQ, Onxeo, The Longevity Labs, Inzen, Sotio, Promontory, Noxopharm, EduCom and the Luke Heller TECPR2 Foundation, and holds Promontory stock options. All other authors declare no conflicts of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
