

Immune evasion and provocation by Mycobacterium tuberculosis
- PMID: 35879556
- PMCID: PMC9310001
- DOI: 10.1038/s41579-022-00763-4
Immune evasion and provocation by Mycobacterium tuberculosis
Abstract
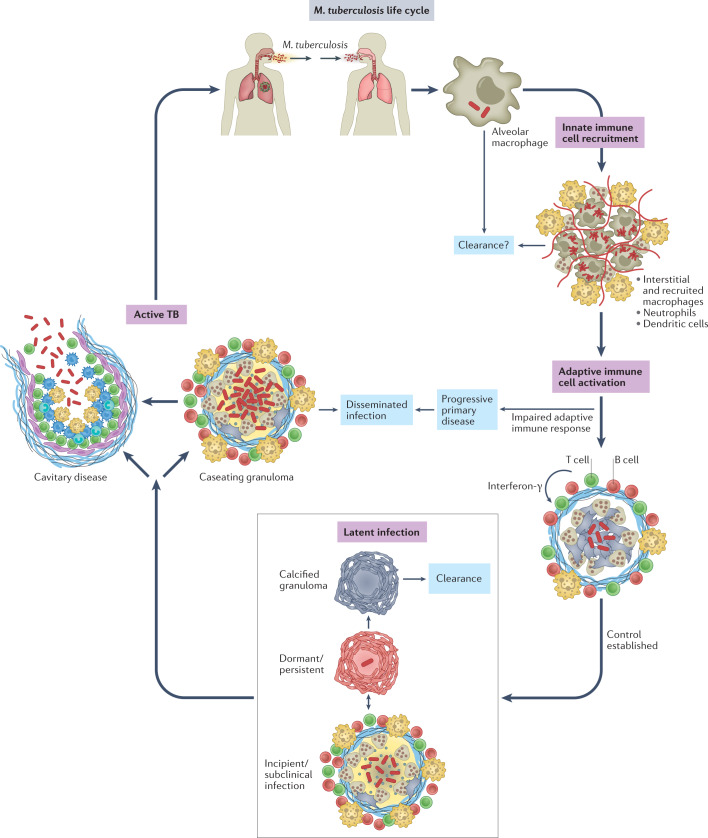
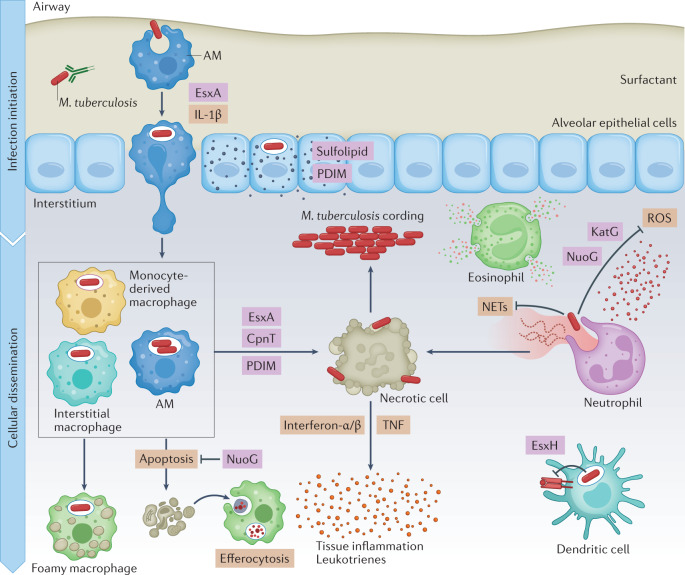
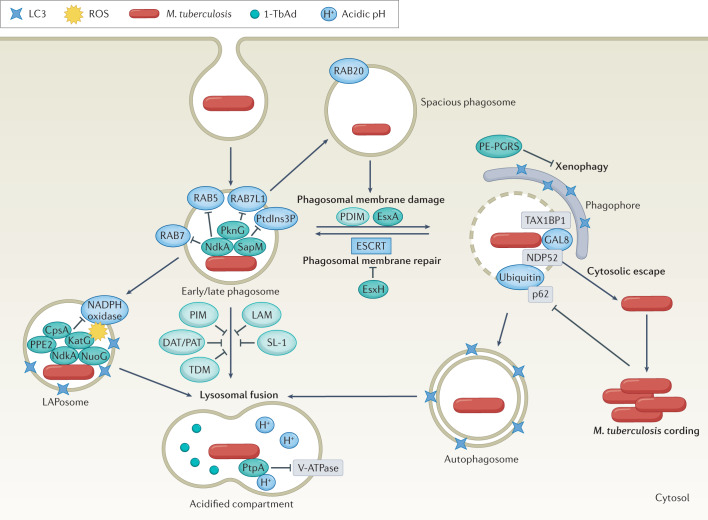
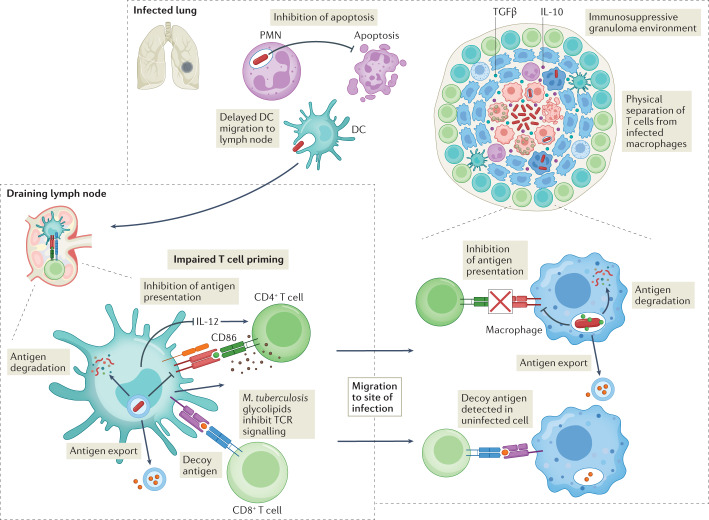
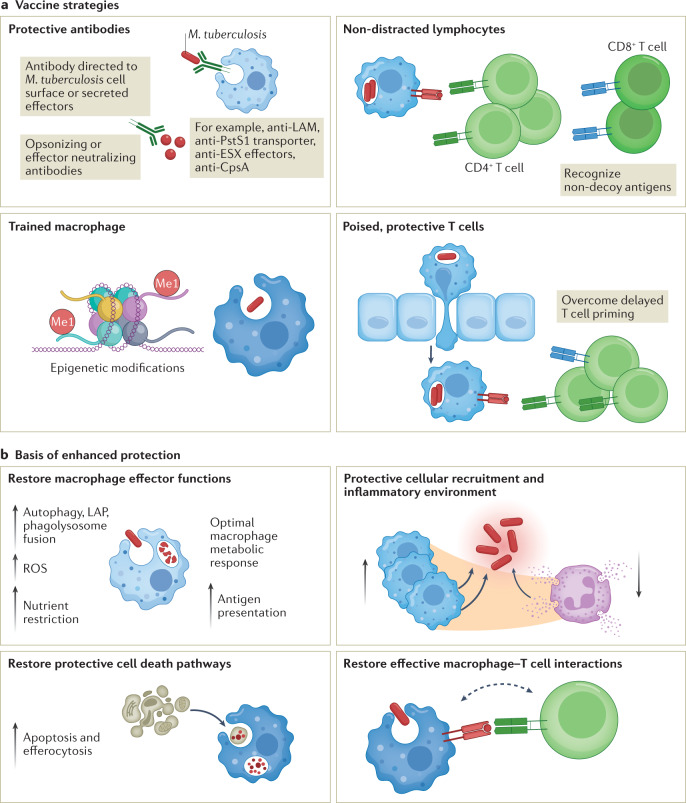
Mycobacterium tuberculosis, the causative agent of tuberculosis, has infected humans for millennia. M. tuberculosis is well adapted to establish infection, persist in the face of the host immune response and be transmitted to uninfected individuals. Its ability to complete this infection cycle depends on it both evading and taking advantage of host immune responses. The outcome of M. tuberculosis infection is often a state of equilibrium characterized by immunological control and bacterial persistence. Recent data have highlighted the diverse cell populations that respond to M. tuberculosis infection and the dynamic changes in the cellular and intracellular niches of M. tuberculosis during the course of infection. M. tuberculosis possesses an arsenal of protein and lipid effectors that influence macrophage functions and inflammatory responses; however, our understanding of the role that specific bacterial virulence factors play in the context of diverse cellular reservoirs and distinct infection stages is limited. In this Review, we discuss immune evasion and provocation by M. tuberculosis during its infection cycle and describe how a more detailed molecular understanding is crucial to enable the development of novel host-directed therapies, disease biomarkers and effective vaccines.
© 2022. Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
