

Phylogenomic Analyses of 2,786 Genes in 158 Lineages Support a Root of the Eukaryotic Tree of Life between Opisthokonts and All Other Lineages
- PMID: 35880421
- PMCID: PMC9366629
- DOI: 10.1093/gbe/evac119
Phylogenomic Analyses of 2,786 Genes in 158 Lineages Support a Root of the Eukaryotic Tree of Life between Opisthokonts and All Other Lineages
Abstract
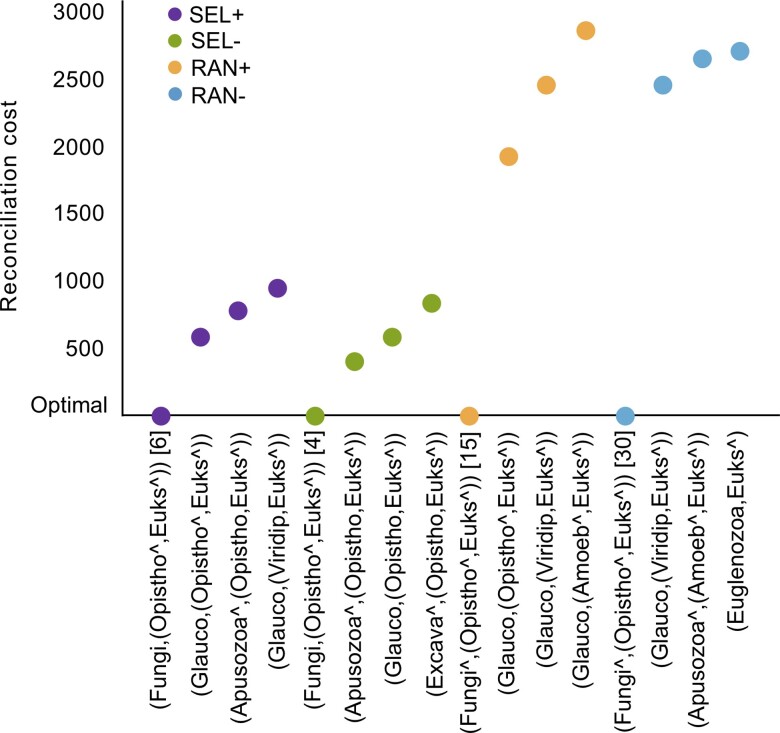
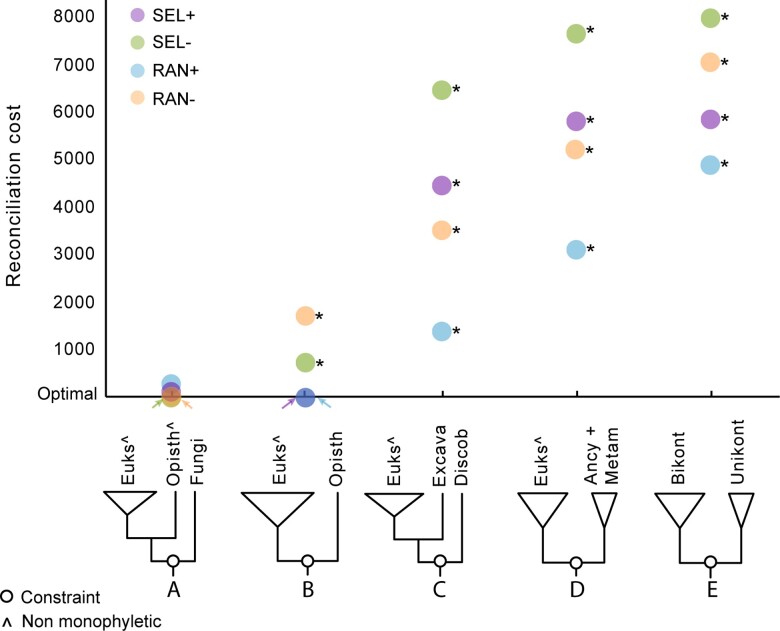
Advances in phylogenomics and high-throughput sequencing have allowed the reconstruction of deep phylogenetic relationships in the evolution of eukaryotes. Yet, the root of the eukaryotic tree of life remains elusive. The most popular hypothesis in textbooks and reviews is a root between Unikonta (Opisthokonta + Amoebozoa) and Bikonta (all other eukaryotes), which emerged from analyses of a single-gene fusion. Subsequent, highly cited studies based on concatenation of genes supported this hypothesis with some variations or proposed a root within Excavata. However, concatenation of genes does not consider phylogenetically-informative events like gene duplications and losses. A recent study using gene tree parsimony (GTP) suggested the root lies between Opisthokonta and all other eukaryotes, but only including 59 taxa and 20 genes. Here we use GTP with a duplication-loss model in a gene-rich and taxon-rich dataset (i.e., 2,786 gene families from two sets of 155 and 158 diverse eukaryotic lineages) to assess the root, and we iterate each analysis 100 times to quantify tree space uncertainty. We also contrasted our results and discarded alternative hypotheses from the literature using GTP and the likelihood-based method SpeciesRax. Our estimates suggest a root between Fungi or Opisthokonta and all other eukaryotes; but based on further analysis of genome size, we propose that the root between Opisthokonta and all other eukaryotes is the most likely.
Keywords: gene duplication; gene loss; gene tree parsimony; gene tree–species tree reconciliation; maximum likelihood; phylogenomics; root of eukaryotes.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
