

[Late onset Pompe disease: an analysis of 19 patients from Mexico]
- PMID: 35880963
- PMCID: PMC10280748
- DOI: 10.33588/rn.7505.2022227
[Late onset Pompe disease: an analysis of 19 patients from Mexico]
Abstract
Introduction: Pompe disease (PD) is a rare metabolic myopathy with an ample and heterogeneous clinical spectrum, particularly late onset PD (LOPD), which is characterized by appearance at older age and slower disease progression, leading to diagnostic confirmation difficulty and delay.
Aim: To describe the genotype and clinical characteristics of Mexican patients with LOPD.
Material and methods: Clinical information from 19 Mexican patients with LOPD confirmed with enzyme activity and GAA gene analysis was reviewed. Genetic information of our population was crossed with international genetic databases.
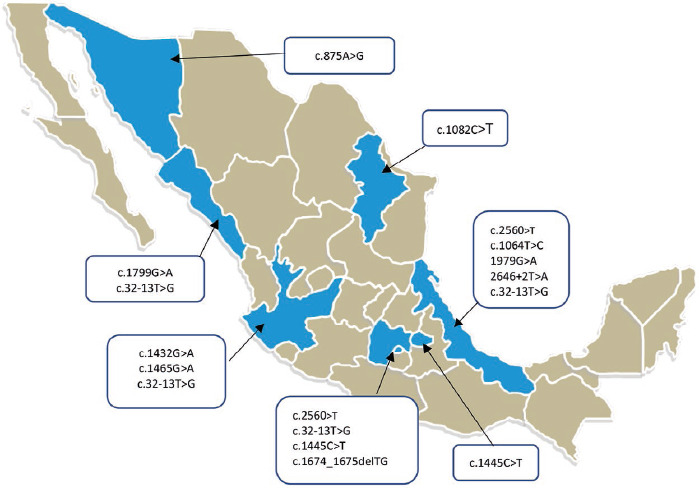
Results: Median age between onset of symptoms and diagnosis was 19 years (range 2-43) and diagnostic confirmation 36 years (range 9-52). Most frequently referred symptoms were proximal axial weakness (n = 17; 89.5%), waddling gait (n = 17; 89.5%) and hyperlordosis (n = 7; 36.8%). Sixteen patients (84.2%) were evaluated with electromyography; a myopathic pattern was reported in 11 (57.8%), but only in 5 patients (26%) paraspinal muscle evaluation was included. The most pathogenic mutations in our group were c.-32-13T>G, c.1799G>A and c.1082C>T.
Conclusions: Similar to other international publications, LOPD in Mexico is clinically heterogeneous; patients may delay years before diagnosis is established. Axial and proximal weakness is the most frequent clinical feature; thus, electromyography with paraspinal muscle evaluation is essential. Except for one, the mutations found in our patients have been previously reported in PD genetic databases.
Title: Enfermedad de Pompe de inicio tardío: análisis de una casuística de 19 pacientes mexicanos.
Introducción. La enfermedad de Pompe es una miopatía metabólica rara con espectro clínico heterogéneo, especialmente la de inicio tardío, cuya sintomatología es de progresión más lenta y representa un gran reto diagnóstico. Objetivo. Describir el genotipo y las características clínicas de pacientes mexicanos con Pompe de inicio tardío (LOPD). Material y métodos. Se incluyó a 19 pacientes mexicanos con LOPD confirmada mediante actividad enzimática y estudio molecular del gen GAA. Se evaluaron datos clínicos y se revisaron las mutaciones en bases de datos genómicas. Resultados. La mediana de edad de inicio de los síntomas fue de 19 años (rango: 2-43 años), y la edad de diagnóstico, de 36 años (rango: 9-52 años). Los síntomas más frecuentes fueron debilidad axial y proximal (n = 17; 89,5%), marcha basculante (n = 17; 89,5%) e hiperlordosis (n = 7; 36,8%). A 16 pacientes (84,2%) se les realizó electromiografía; 11 (57,8%) describieron patrón miopático y sólo en cinco pacientes (26%) se incluyó la valoración de los músculos paraespinales. Las variantes patogénicas más frecuentes en nuestra casuística fueron c.-32-13T>G, c.1799G>A y c.1082C>T. Conclusiones. Parecido a lo comunicado en publicaciones internacionales, la LOPD en México es clínicamente heterogénea; los pacientes pueden tardar años en llegar al diagnóstico. La debilidad muscular axial y proximal es el dato clínico más frecuente, por lo que la electromiografía debe incluir valoración de los músculos paraespinales. A excepción de una, las mutaciones encontradas en nuestra serie de casos se encuentran previamente descritas en las bases de datos de enfermedad de Pompe.
Conflict of interest statement
Conflicto de intereses: V.M.M., E.K. y P.F.R.D. trabajan para el departamento médico de enfermedades raras de Sanofi Genzyme. Su participación fue mediante el análisis de información y apoyo en redacción, sin participar en la discusión.
Figures
Similar articles
-
Mutational spectrum and genotype-phenotype correlation in Mexican patients with infantile-onset and late-onset Pompe disease.Mol Genet Genomic Med. 2024 Jul;12(7):e2480. doi: 10.1002/mgg3.2480. Mol Genet Genomic Med. 2024. PMID: 38958145 Free PMC article.
-
Early-onset of symptoms and clinical course of Pompe disease associated with the c.-32-13 T > G variant.Mol Genet Metab. 2019 Feb;126(2):106-116. doi: 10.1016/j.ymgme.2018.08.009. Epub 2018 Aug 23. Mol Genet Metab. 2019. PMID: 30655185 Free PMC article.
-
[GAA gene variants and genotype-phenotype correlations in patients with glycogen storage disease type Ⅱ].Zhonghua Er Ke Za Zhi. 2021 Mar 2;59(3):189-194. doi: 10.3760/cma.j.cn112140-20200710-00710. Zhonghua Er Ke Za Zhi. 2021. PMID: 33657692 Chinese.
-
Newborn screening for Pompe disease in Japan: report and literature review of mutations in the GAA gene in Japanese and Asian patients.J Hum Genet. 2019 Aug;64(8):741-755. doi: 10.1038/s10038-019-0603-7. Epub 2019 May 10. J Hum Genet. 2019. PMID: 31076647 Review.
-
Pompe disease: literature review and case series.Neurol Clin. 2014 Aug;32(3):751-76, ix. doi: 10.1016/j.ncl.2014.04.010. Neurol Clin. 2014. PMID: 25037089 Free PMC article. Review.
Cited by
-
Mutational spectrum and genotype-phenotype correlation in Mexican patients with infantile-onset and late-onset Pompe disease.Mol Genet Genomic Med. 2024 Jul;12(7):e2480. doi: 10.1002/mgg3.2480. Mol Genet Genomic Med. 2024. PMID: 38958145 Free PMC article.
-
[Juvenile Pompe disease: Undescribed genotype. First report in Quintana Roo].Rev Med Inst Mex Seguro Soc. 2024 Jan 8;62(1):1-5. doi: 10.5281/zenodo.10278165. Rev Med Inst Mex Seguro Soc. 2024. PMID: 39110956 Free PMC article. Spanish.
References
-
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med 2006;8:267-88. - PMC - PubMed
- Kishnani PS, Steiner RD, Bali D, Berger K, Byrne BJ, Case LE, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006;8:267–88. - PMC - PubMed
-
- Dubroysky A, Fulgenzi A, Amartino H, Carlés D, Corderi J, De Vito E, et al. Consenso argentino para el diagnóstico, seguimiento y tratamiento de la enfermedad de Pompe. Neurología Argentina 2014;6:96-113.
- Dubroysky A, Fulgenzi A, Amartino H, Carlés D, Corderi J, De Vito E, et al. Consenso argentino para el diagnóstico, seguimiento y tratamiento de la enfermedad de Pompe. Neurología Argentina. 2014;6:96–113.
-
- Barba-Romero MA, Barrot E, Bautista-Lorite J, Gutierrez-Rivas E, Illa I, Jimenez LM, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol 2012;54:497-507. - PubMed
- Barba-Romero MA, Barrot E, Bautista-Lorite J, Gutierrez-Rivas E, Illa I, Jimenez LM, et al. Clinical guidelines for late-onset Pompe disease. Rev Neurol. 2012;54:497–507. - PubMed
-
- Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of alpha-glucosidase activities in dried blood spots. Mol Genet Metab 2007;90:449-52. - PubMed
- Kallwass H, Carr C, Gerrein J, Titlow M, Pomponio R, Bali D, et al. Rapid diagnosis of late-onset Pompe disease by fluorometric assay of alpha-glucosidase activities in dried blood spots. Mol Genet Metab. 2007;90:449–52. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
