

Low abundance members of the gut microbiome exhibit high immunogenicity
- PMID: 35881003
- PMCID: PMC9331198
- DOI: 10.1080/19490976.2022.2104086
Low abundance members of the gut microbiome exhibit high immunogenicity
Abstract
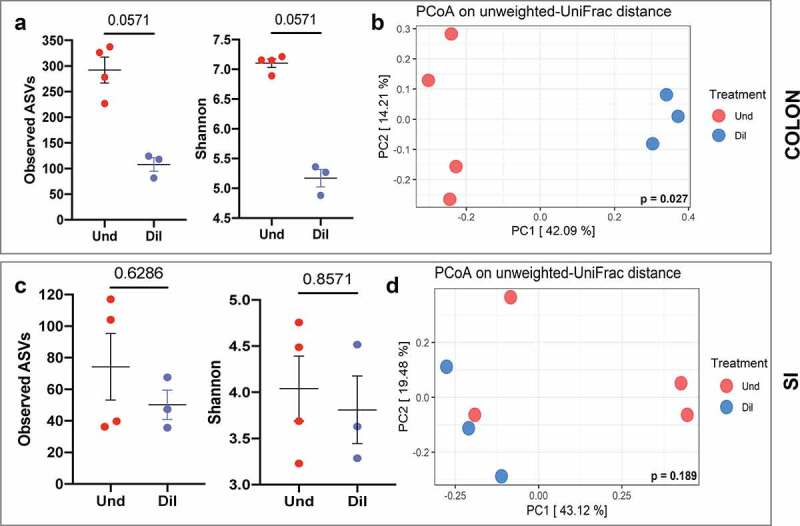
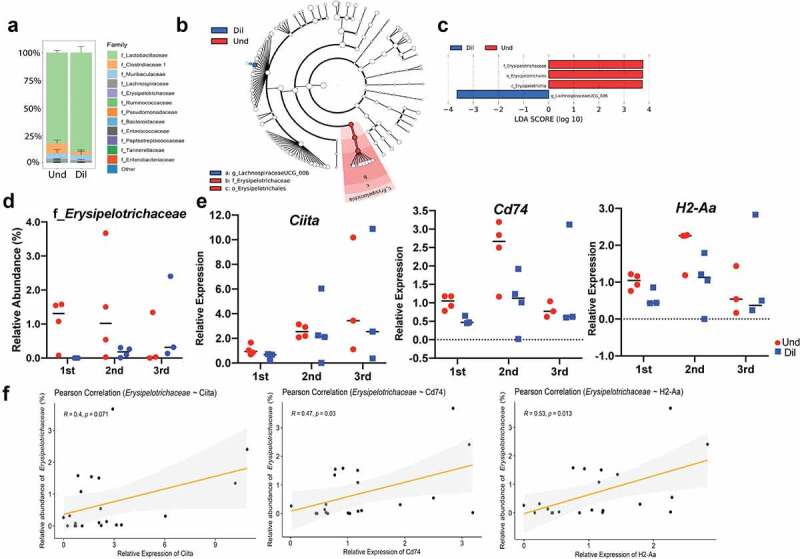
Studies identifying bacterial members that dictate host phenotype have focused mainly on the dominant members, and the role of low abundance microbes in determining host phenotypes and pathogenesis of diseases remains unexplored. In this study, we compared the gut bacterial community of mice with wide-ranging microbial exposure to determine if low abundance bacteria vary based on microbial exposure or remain consistent. We noted that similar to the high abundance bacterial community, a core community of low abundance bacteria made up a significant portion of the gut microbiome irrespective of microbial exposure. To determine the role of low abundance bacteria in regulating community composition and host gene expression, we devised a microbiome dilution strategy to "delete" out low abundance bacteria and engrafted the diluted microbiomes into germ-free mice. Our approach successfully excluded low abundance bacteria from small and large intestinal bacterial communities and induced global changes in microbial community composition in the large intestine. Gene expression analysis of intestinal tissue revealed that loss of low abundance bacteria resulted in a drastic reduction in expression of multiple genes involved MHCII antigen presentation pathway and T-cell cytokine production in the small intestine. The effect of low abundance bacteria on MHCII expression was found to be specific to the intestinal epithelium at an early timepoint post-colonization and correlated with bacteria belonging to the family Erysipelotrichaceae. We conclude that low abundance bacteria have a significantly higher immuno-stimulatory effect compared to dominant bacteria and are thus potent drivers of early immune education in the gut.
Keywords: Erysipelotrichaceae; Gut microbiome; MHC class II; immune education; low abundance bacteria.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials