

Tracking Energy Transfer across a Platinum Center
- PMID: 35881911
- PMCID: PMC9358659
- DOI: 10.1021/acs.jpca.2c02017
Tracking Energy Transfer across a Platinum Center
Abstract
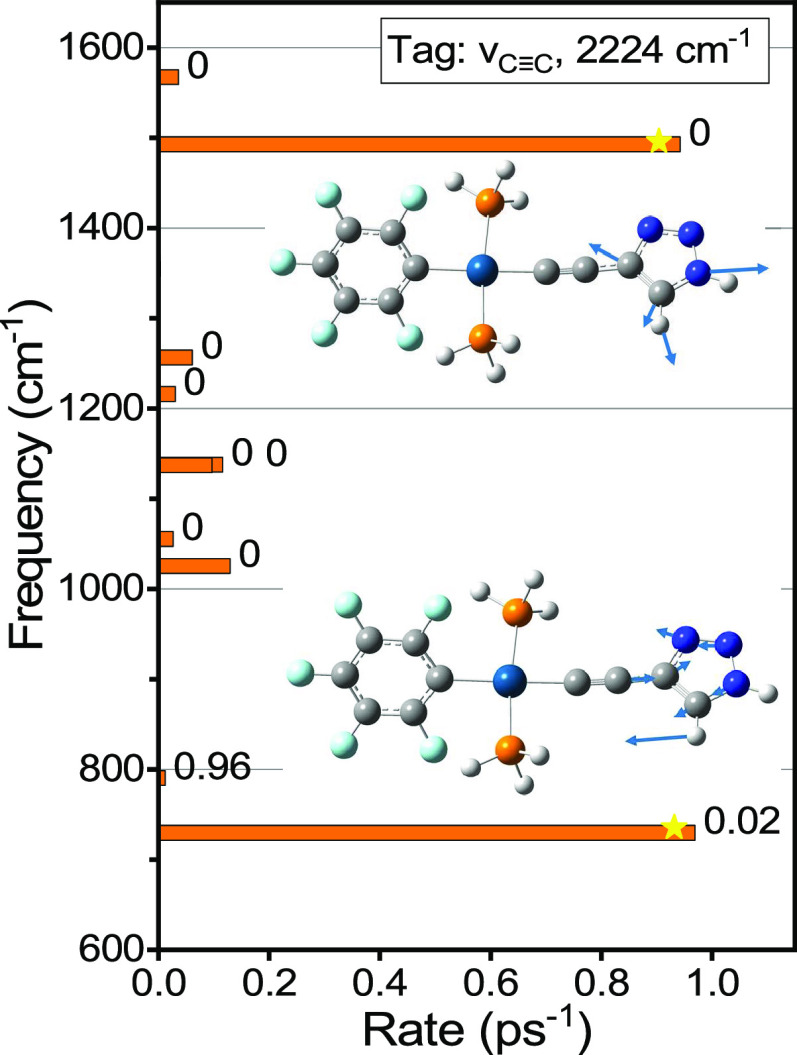
Rigid, conjugated alkyne bridges serve as important components in various transition-metal complexes used for energy conversion, charge separation, sensing, and molecular electronics. Alkyne stretching modes have potential for modulating charge separation in donor-bridge-acceptor compounds. Understanding the rules of energy relaxation and energy transfer across the metal center in such compounds can help optimize their electron transfer switching properties. We used relaxation-assisted two-dimensional infrared spectroscopy to track energy transfer across metal centers in platinum complexes featuring a triazole-terminated alkyne ligand of two or six carbons, a perfluorophenyl ligand, and two tri(p-tolyl)phosphine ligands. Comprehensive analyses of waiting-time dynamics for numerous cross and diagonal peaks were performed, focusing on coherent oscillation, energy transfer, and cooling parameters. These observables augmented with density functional theory computations of vibrational frequencies and anharmonic force constants enabled identification of different functional groups of the compounds. Computations of vibrational relaxation pathways and mode couplings were performed, and two regimes of intramolecular energy redistribution are described. One involves energy transfer between ligands via high-frequency modes; the transfer is efficient only if the modes involved are delocalized over both ligands. The energy transport pathways between the ligands are identified. Another regime involves redistribution via low-frequency delocalized modes, which does not lead to interligand energy transport.
Conflict of interest statement
The authors declare no competing financial interest.
Figures














References
-
- Dembinski R.; Bartik T.; Bartik B.; Jaeger M.; Gladysz J. A. Toward Metal-Capped One-Dimensional Carbon Allotropes: Wirelike C6–C20 Polyynediyl Chains That Span Two Redox-Active (η5-C5Me5)Re (NO)(PPh3) Endgroups. J. Am. Chem. Soc. 2000, 122, 810–822. 10.1021/ja992747z. - DOI
-
- Zheng Q.; Bohling J. C.; Peters T. B.; Frisch A. C.; Hampel F.; Gladysz J. A. A Synthetic Breakthrough into an Unanticipated Stability Regime: A Series of Isolable Complexes in which C6, C8, C10, C12, C16, C20, C24, and C28 Polyynediyl Chains Span Two Platinum Atoms. Chem. – Eur. J. 2006, 12, 6486–6505. 10.1002/chem.200600615. - DOI - PubMed
-
- Gauthier S.; Weisbach N.; Bhuvanesh N.; Gladysz J. A. “Click” Chemistry in Metal Coordination Spheres: Copper(I)-Catalyzed 3+2 Cycloadditions of Benzyl Azide and Platinum Polyynyl Complexes trans-(C6F5)(p-tol3P)2Pt(C≡C)nH (n = 2–6). Organometallics 2009, 28, 5597–5599. 10.1021/om900680q. - DOI
-
- Zheng Q.; Schneider J. F.; Amini H.; Hampel F.; Gladysz J. A. Wire like diplatinum, triplatinum, and tetraplatinum complexes featuring X[PtC≡CC≡CC≡CC≡C]mPtX segments; iterative syntheses and functionalization for measurements of single molecule properties. Dalton Trans. 2019, 48, 5800–5816. 10.1039/c9dt00870e. - DOI - PubMed
-
- Weisbach N.; Kuhn H.; Amini H.; Ehnbom A.; Hampel F.; Reibenspies J. H.; Hall M. B.; Gladysz J. A. Triisopropylsilyl (TIPS) Alkynes as Building Blocks for Syntheses of Platinum Triisopropylsilylpolyynyl and Diplatinum Polyynediyl Complexes. Organometallics 2019, 38, 3294–3310. 10.1021/acs.organomet.9b00368. - DOI
LinkOut - more resources
Full Text Sources
