

HINT1 neuropathy: Expanding the genotype and phenotype spectrum
- PMID: 35882622
- PMCID: PMC9805104
- DOI: 10.1111/cge.14198
HINT1 neuropathy: Expanding the genotype and phenotype spectrum
Abstract
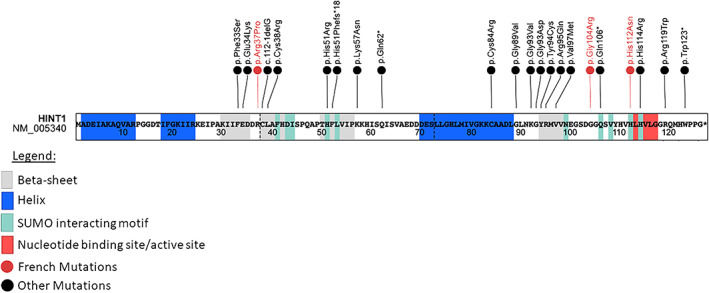
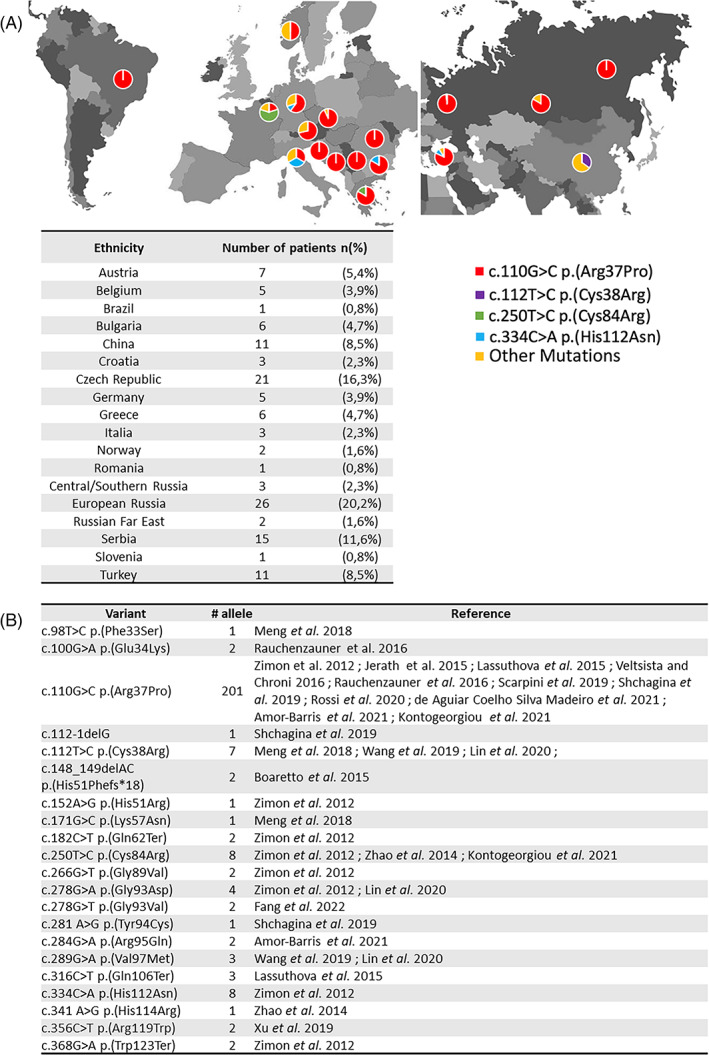
Inherited peripheral neuropathy (IPN) is a heterogeneous group of disorders due to pathogenic variation in more than 100 genes. In 2012, the first cases of IPN associated with HINT1 pathogenic variations were described in 33 families sharing the same phenotype characterized by an axonal neuropathy with neuromyotonia and autosomal recessive inheritance (NMAN: OMIM #137200). Histidine Triad Nucleotide Binding Protein 1 regulates transcription, cell-cycle control, and is possibly involved in neuropsychiatric pathophysiology. Herein, we report seven French patients with NMAN identified by Next Generation Sequencing. We conducted a literature review and compared phenotypic and genotypic features with our cohort. We identified a new HINT1 pathogenic variation involved in NMAN: c.310G>C p.(Gly104Arg). This cohort is comparable with literature data regarding age of onset (7,4yo), neuronal involvement (sensorimotor 3/7 and motor pure 4/7), and skeletal abnormalities (scoliosis 3/7, feet anomalies 6/7). We expand the phenotypic spectrum of HINT1-related neuropathy by describing neurodevelopmental or psychiatric features in six out of seven individuals such as generalized anxiety disorder (GAD), obsessive-compulsive disorder (OCD), mood disorder and attention deficit hyperactivity disorder (ADHD). However, only 3/128 previously described patients had neuropsychiatric symptomatology or neurodevelopmental disorder. These features could be part of HINT1-related disease, and we should further study the clinical phenotype of the patients.
Keywords: ARAN-NM; Charcot-Marie-tooth disease; France; HINT1; NMAN; Neuromyotonia; Peripheral neuropathy.
© 2022 The Authors. Clinical Genetics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures
References
-
- Schöler J, Ferralli J, Thiry S, Chiquet‐Ehrismann R. The intracellular domain of teneurin‐1 induces the activity of microphthalmia‐associated transcription factor (MITF) by binding to transcriptional repressor HINT1. J Biol Chem. 2015;290(13):8154‐8165. doi:10.1074/jbc.M114.615922 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
Research Materials
