

Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer
- PMID: 35882862
- PMCID: PMC9325712
- DOI: 10.1038/s41467-022-31963-4
Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer
Erratum in
-
Author Correction: Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer.Nat Commun. 2022 Aug 8;13(1):4640. doi: 10.1038/s41467-022-32459-x. Nat Commun. 2022. PMID: 35941179 Free PMC article. No abstract available.
Abstract
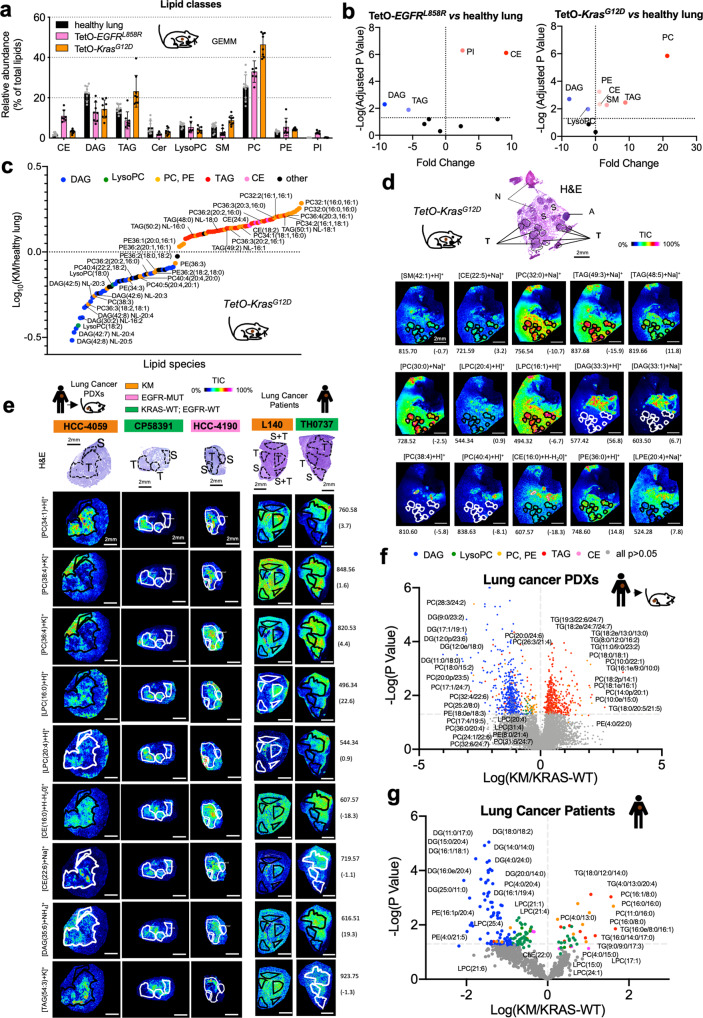
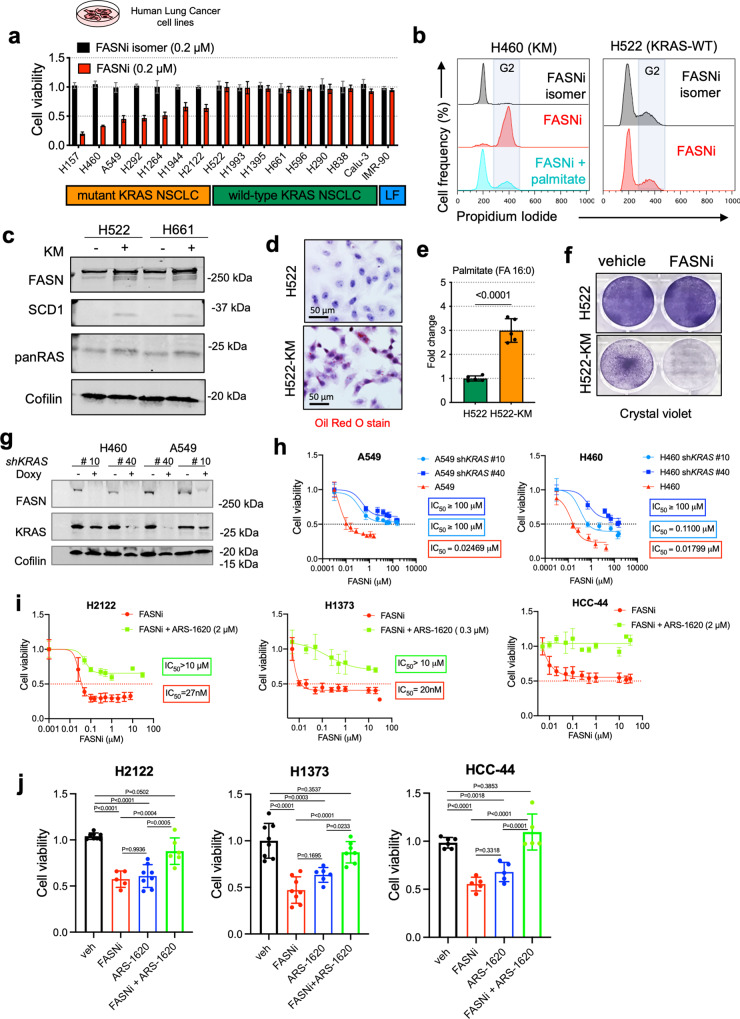
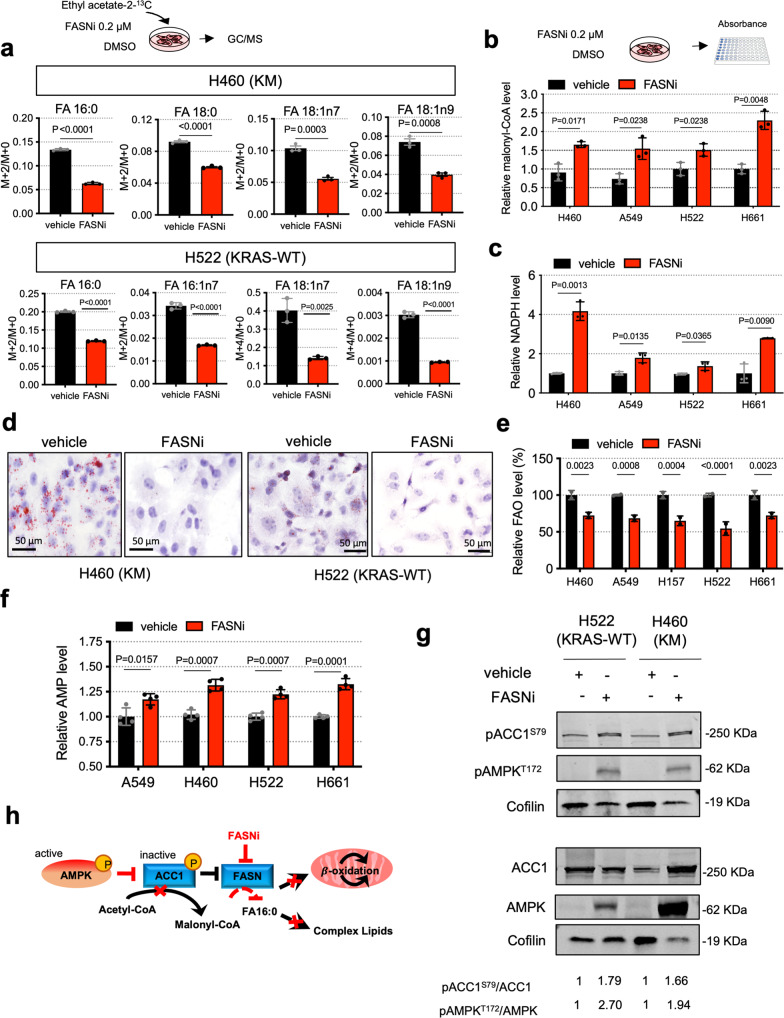
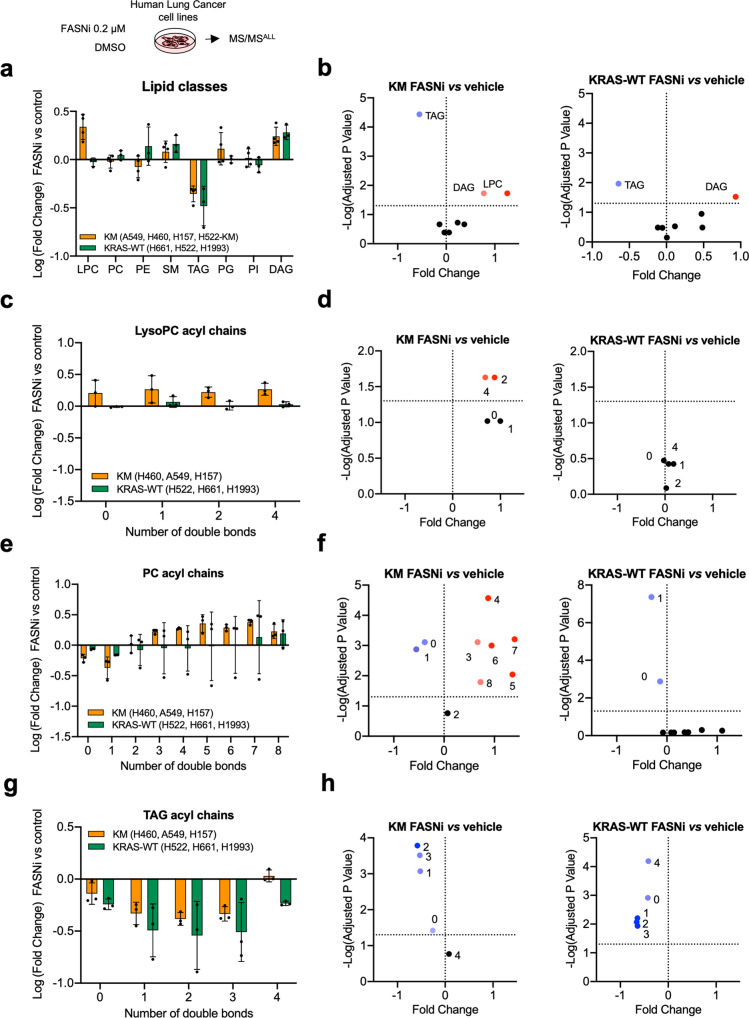
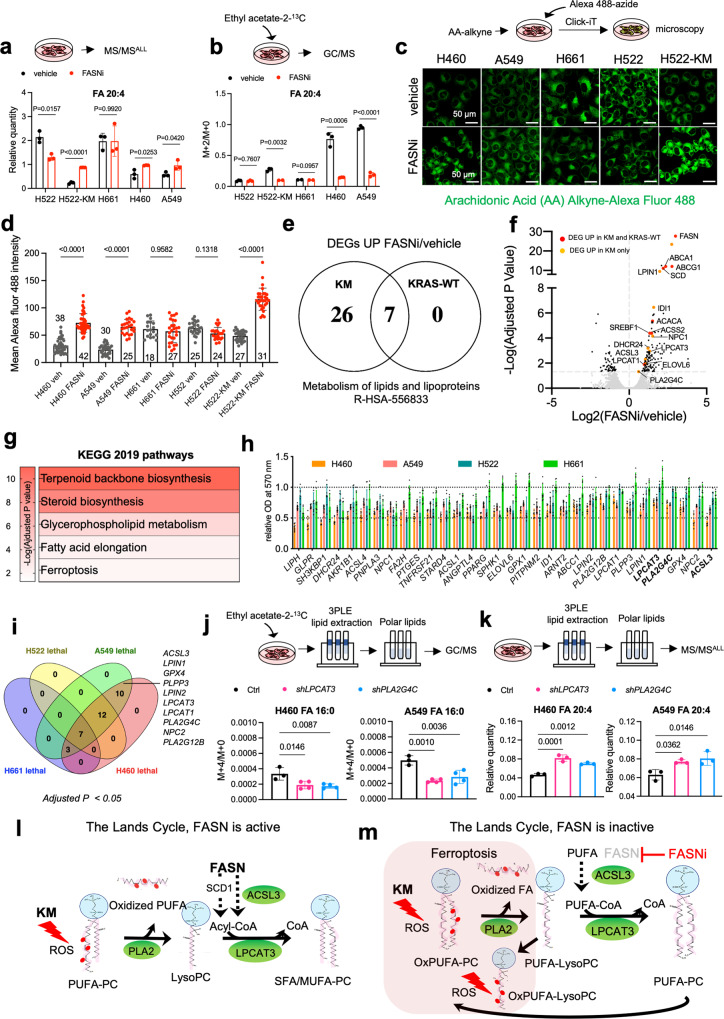
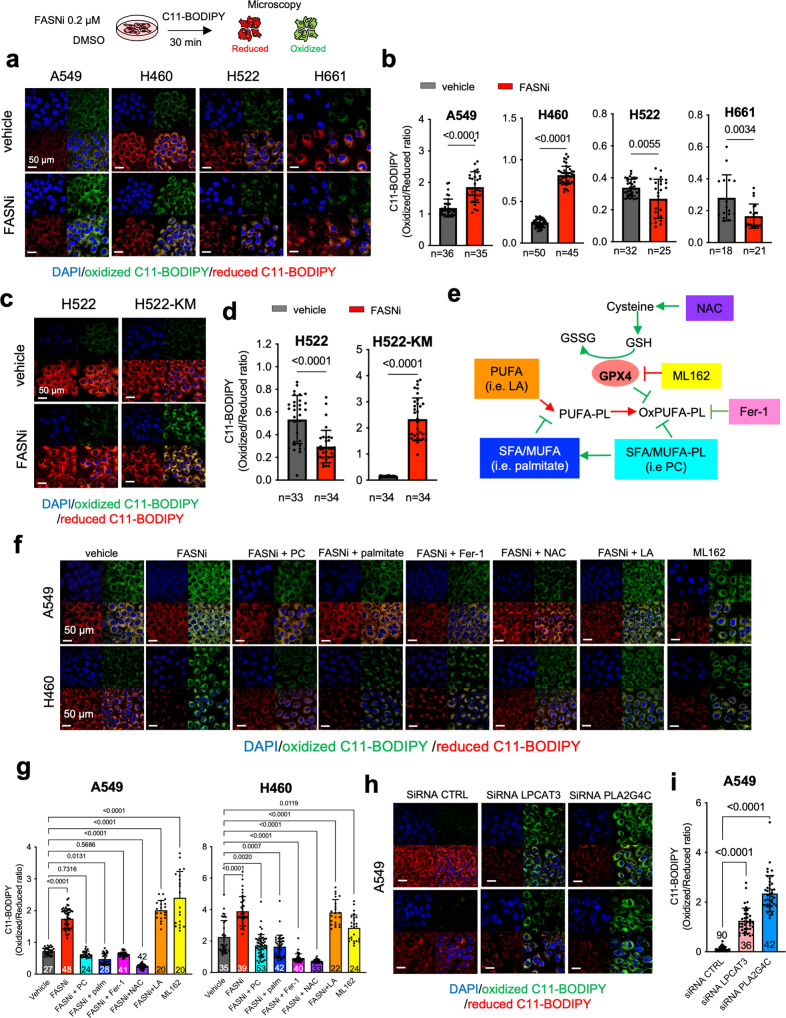
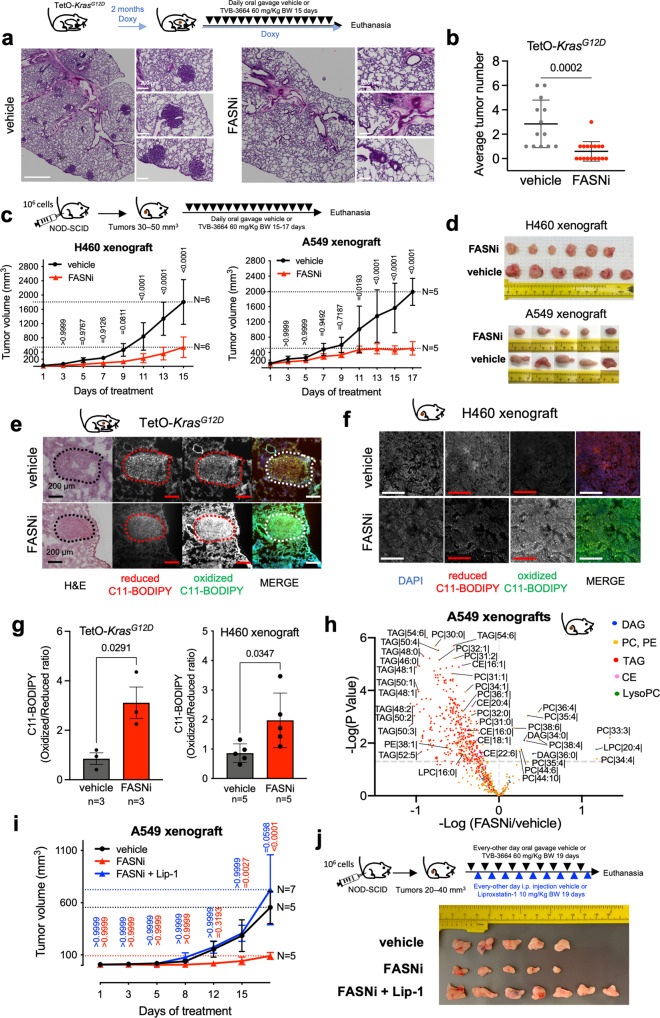
Mutant KRAS (KM), the most common oncogene in lung cancer (LC), regulates fatty acid (FA) metabolism. However, the role of FA in LC tumorigenesis is still not sufficiently characterized. Here, we show that KMLC has a specific lipid profile, with high triacylglycerides and phosphatidylcholines (PC). We demonstrate that FASN, the rate-limiting enzyme in FA synthesis, while being dispensable in EGFR-mutant or wild-type KRAS LC, is required for the viability of KMLC cells. Integrating lipidomic, transcriptomic and functional analyses, we demonstrate that FASN provides saturated and monounsaturated FA to the Lands cycle, the process remodeling oxidized phospholipids, such as PC. Accordingly, blocking either FASN or the Lands cycle in KMLC, promotes ferroptosis, a reactive oxygen species (ROS)- and iron-dependent cell death, characterized by the intracellular accumulation of oxidation-prone PC. Our work indicates that KM dictates a dependency on newly synthesized FA to escape ferroptosis, establishing a targetable vulnerability in KMLC.
© 2022. The Author(s).
Conflict of interest statement
G.K. is CEO and Chief Scientific Officer at Sagimet Biosciences where TVB-3664 has been developed. The other authors declare no competing interests.
Figures
References
-
- McCormick, F. Progress in targeting RAS with small molecule drugs. Biochem. J. 10.1042/BCJ20170441 (2019). - PubMed
-
- Canon, J. et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature10.1038/s41586-019-1694-1 (2019). - PubMed
-
- Hallin, J. et al. The KRASG12C inhibitor MRTX849 provides insight toward therapeutic susceptibility of KRAS-mutant cancers in mouse models and patients. Cancer Discov. 10.1158/2159-8290.CD-19-1167 (2020). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
