

A systematic analysis of splicing variants identifies new diagnoses in the 100,000 Genomes Project
- PMID: 35883178
- PMCID: PMC9327385
- DOI: 10.1186/s13073-022-01087-x
A systematic analysis of splicing variants identifies new diagnoses in the 100,000 Genomes Project
Abstract
Background: Genomic variants which disrupt splicing are a major cause of rare genetic diseases. However, variants which lie outside of the canonical splice sites are difficult to interpret clinically. Improving the clinical interpretation of non-canonical splicing variants offers a major opportunity to uplift diagnostic yields from whole genome sequencing data.
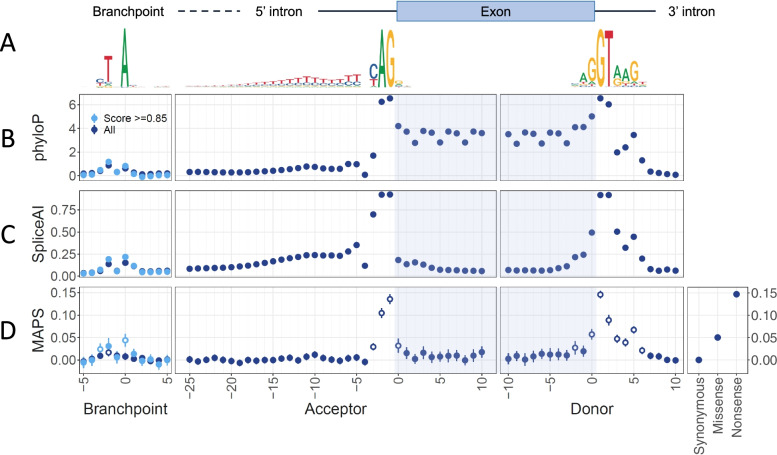
Methods: Here, we examine the landscape of splicing variants in whole-genome sequencing data from 38,688 individuals in the 100,000 Genomes Project and assess the contribution of non-canonical splicing variants to rare genetic diseases. We use a variant-level constraint metric (the mutability-adjusted proportion of singletons) to identify constrained functional variant classes near exon-intron junctions and at putative splicing branchpoints. To identify new diagnoses for individuals with unsolved rare diseases in the 100,000 Genomes Project, we identified individuals with de novo single-nucleotide variants near exon-intron boundaries and at putative splicing branchpoints in known disease genes. We identified candidate diagnostic variants through manual phenotype matching and confirmed new molecular diagnoses through clinical variant interpretation and functional RNA studies.
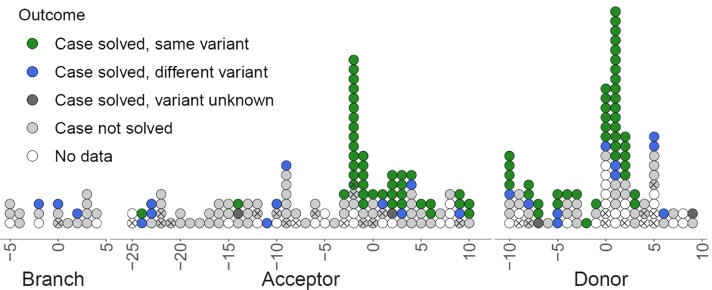
Results: We show that near-splice positions and splicing branchpoints are highly constrained by purifying selection and harbour potentially damaging non-coding variants which are amenable to systematic analysis in sequencing data. From 258 de novo splicing variants in known rare disease genes, we identify 35 new likely diagnoses in probands with an unsolved rare disease. To date, we have confirmed a new diagnosis for six individuals, including four in whom RNA studies were performed.
Conclusions: Overall, we demonstrate the clinical value of examining non-canonical splicing variants in individuals with unsolved rare diseases.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
-
- International Rare Diseases Research Consortium. Policies and guidelines. (2013). Available at: https://irdirc.org/about-us/policies-guidelines/. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
