

Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature
- PMID: 35884482
- PMCID: PMC9322180
- DOI: 10.3390/cancers14143421
Genetics in Familial Intrahepatic Cholestasis: Clinical Patterns and Development of Liver and Biliary Cancers: A Review of the Literature
Abstract
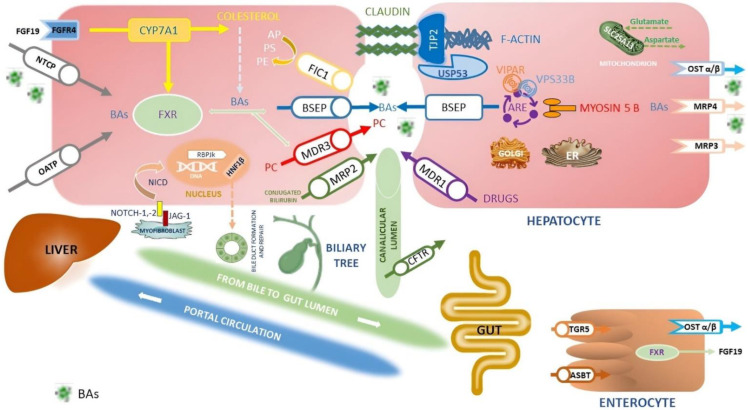
The family of inherited intrahepatic cholestasis includes autosomal recessive cholestatic rare diseases of childhood involved in bile acids secretion or bile transport defects. Specific genetic pathways potentially cause many otherwise unexplained cholestasis or hepatobiliary tumours in a healthy liver. Lately, next-generation sequencing and whole-exome sequencing have improved the diagnostic procedures of familial intrahepatic cholestasis (FIC), as well as the discovery of several genes responsible for FIC. Moreover, mutations in these genes, even in the heterozygous status, may be responsible for cryptogenic cholestasis in both young and adults. Mutations in FIC genes can influence serum and hepatic levels of bile acids. Experimental studies on the NR1H4 gene have shown that high bile acids concentrations cause excessive production of inflammatory cytokines, resistance to apoptosis, and increased cell regeneration, all risk conditions for developing hepatocellular carcinoma (HCC) and cholangiocarcinoma (CCA). NR1H4 gene encodes farnesoid X-activated receptor having a pivotal role in bile salts synthesis. Moreover, HCC and CCA can emerge in patients with several FIC genes such as ABCB11, ABCB4 and TJP2. Herein, we reviewed the available data on FIC-related hepatobiliary cancers, reporting on genetics to the pathophysiology, the risk factors and the clinical presentation.
Keywords: Alagille syndrome; bile acids; cholangiocarcinoma; gallbladder cancer; hepatobiliary cancers; hepatocellular carcinoma; liver transplantation; microbiota; next-generation sequencing; progressive familial intrahepatic cholestasis.
Conflict of interest statement
Fabio Piscaglia has the following conflicts of interest to disclose. Consulting or lecture fees in the last two years from: Astrazeneca, Bayer, Bracco, EISAI, ESAOTE, Exact Sciences, IPSEN, MSD, Roche, Samsung, and Tiziana Life Sciences. The other authors do not declare conflicts of interest.
Figures
References
-
- Vitale G., Gitto S., Raimondi F., Mattiaccio A., Mantovani V., Vukotic R., D’Errico A., Seri M., Russell R.B., Andreone P. Cryptogenic cholestasis in young and adults: ATP8B1, ABCB11, ABCB4, and TJP2 gene variants analysis by high-throughput sequencing. J. Gastroenterol. 2018;53:945–958. doi: 10.1007/s00535-017-1423-1. - DOI - PubMed
-
- Yamada S., Takashina Y., Watanabe M., Nagamine R., Saito Y., Kamada N., Saito H. Bile acid metabolism regulated by the gut microbiota promotes non-alcoholic steatohepatitis-associated hepatocellular carcinoma in mice. Oncotarget. 2018;9:9925–9939. doi: 10.18632/oncotarget.24066. - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
